LYSOSOMES
Lysosomes were discovered by Duve and given the name (1955). They are also called suicidal bags. They can be identified as dense, granular membranous sacs capable of digesting various substances. The marker enzyme for this organelle is acid phosphatase.
The size of lysosomes varies from 0.2 to 1.2um. The membrane that encloses lysosomes is single unit structure. Lysosomal liquid is acidic in pH (4.8) while cytosol pH is ~7.2. It maintains such low pH by pumping protons (H+) across into Lysosomal sac. The number also varies from cell to cell and it depends upon the metabolic state of the cells. Yeast cells under glucose repression produce large number of lysosomes which autophagically digest mitochondria. Similarly during metamorphosis of tadpole into young frog a great number of lysosomes appear in the tail of tadpole and they remain active till the tail is resorbed. Lysosomes are classified into primary lysosomes and secondary Lysosomes. The former are directly derived from Golgi membranes and the latter are the products of fusion between primary lysosomes and endocytotic vesicles or phagosomes.

Isolated single lysosome; http://faculty.muhs.edu/
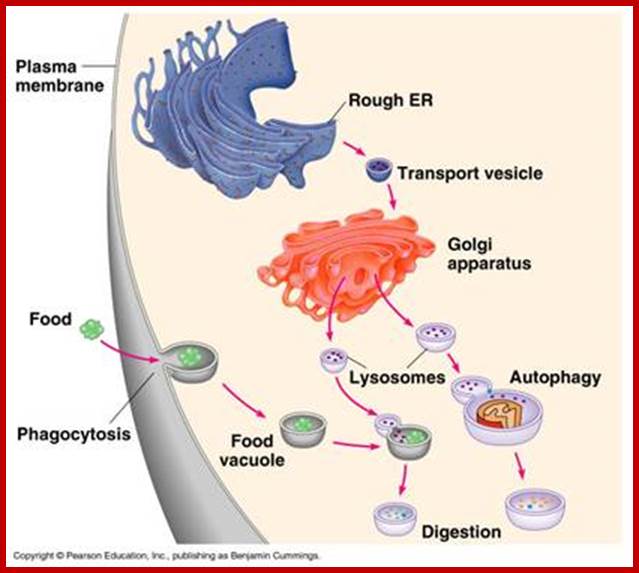
Lysosomes originate within the Golgi apparatus. The digestive enzymes found in the lysosome are manufactured in the rough endoplasmic reticulum (ribosomes bound) of the cell and transported to the trans-golgi membrane where proteins are loaded into vesicles pinched off which is facilitated by Clathrin surface proteins, as primary lysosomes. The lysosomes fuse with early endosome or late endosome to develop into lysosomes
www.dipietrolab.bmb.colostate.edu
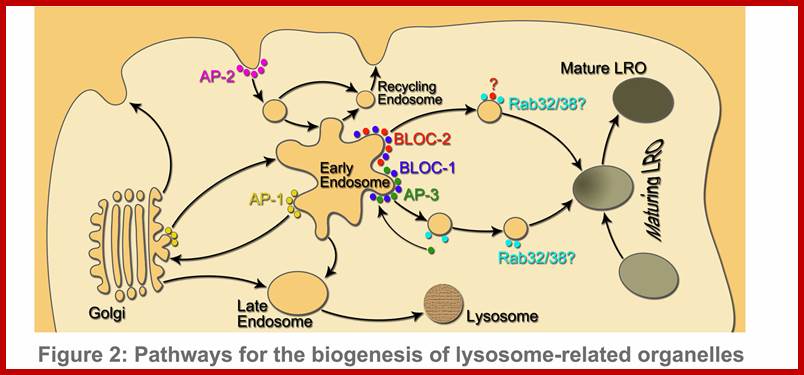
Biogenesis of lysosomes; www.faculty.sau.edu.sa
Structure:
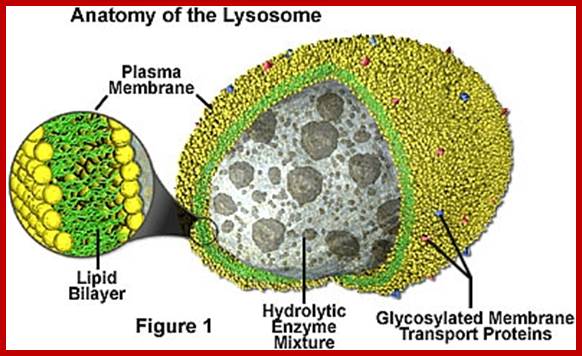
Lysosomes are spherical structures bounded by a single unit membrane within which dense granular enzymatic components are found. Though hydrolysing enzymes are found within the lysosomes, its own membrane is not digested because the membrane is highly modified and resistant to such enzymes; more over the enzymes are rendered inactive by electrostatic binding forces. However lysosomal membranes are sensitive to many labilizers and stabilizers. Vitamins like A, B, K and hormones like progesterone, B-esterdiol and some metal ions like Ag2+, Hg2 +, Cu2 labilize the membranes and allow the enzymes to diffuse out. But cholesterol, cortisones, etc stabilize the membranes. Thus intacellular conditions control and regulate the release of the lysosomal enzymes into cellular fluid.
Lysosomal Enzymes:
More than forty different lysosomal enzymes have been recognized, ex; Nuclease, Acid phosphotase, Lipases, Proteases (Cathapsin A, B, C, D & E), Glycosidase, Lysozyme, Sulfotases and few others. Among them acid phosphotase is a marker enzyme for lysosomes. All the enzymes need not be present in all lysosomes at all times. When lysosomes originate from Golgi bodies they may contain only one set of enzymes but fusion with other lysosomes of different composition produces lysosomes with different enzyme composition (at least 60 of them) -refer to Lysosomal enzyme (https://www.rndsystems.com)
Functions:
Lysosomes release the enzymes on activation by intracellular environment. In plants cells, particularly at the time of seed germination, lysosomal enzymes degrade macromolecules like starch and reserve proteins into glucose and amino acids respectively. In some cases, lysosomal activity results in intercellular or intracellular digestion leading to the formation of schizogenous or lysogenous cavities. Laticiferous cavities found in many Euphorbiaceae and other members are due to the activity of lysosomal enzymes.
In animal cells, the primary lysosomes fuse with food vacuoles derived from phagocytosis and produce phagosomes. After digestion, the residual body is thrown out of the cell by exocytosis. Even the incoming endocytotic vesicles fuse with primary lysosomes into secondary lysosomes. During certain stage of development lysosomes become very active and digest cellular components by autophagy.
Biogenesis of lysosomes
In almost all cases, lysosomes take their origin from golgi-endoplasmic reticulum-lysosomal complex called GERL complex. The RER synthesizes lysosomal proteins on its membrane (polysome bound) then the proteins are transported through the lumen into SER where the membranes are pinched off into vesicles at the cis surface of Golgi body. Lysosomal proteins are marked by mannose-Phosphate. The vesicles fuse with one another into flat membranous sacs. At the lateral regions of Golgi membranes the lysosomal enzymes are segregated and packed. Then many such small primary lysosomes containing similar enzymes or different enzymes fuse with endosomes or endocytotic or phagocytotic vesicle to produce secondary lysosomes.
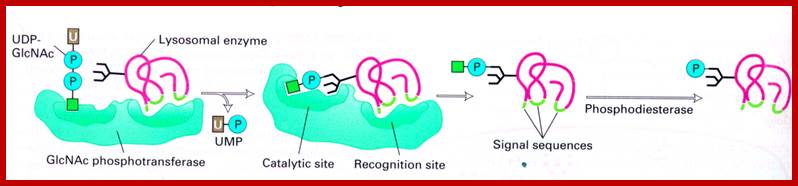
http://cc.scu.edu.cn/
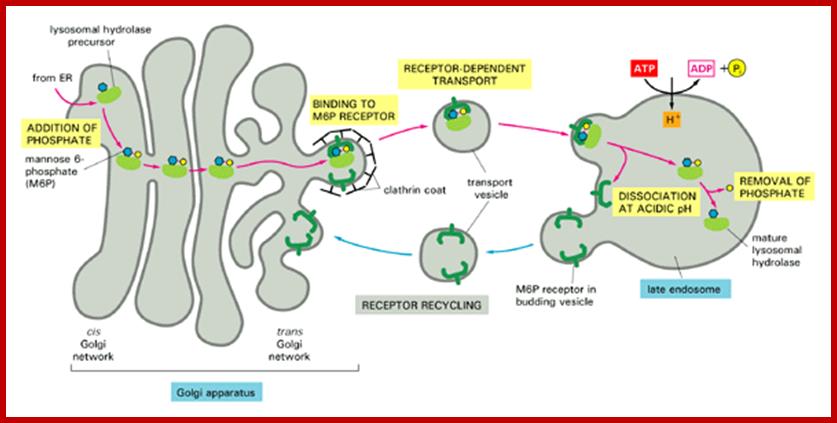
http://bioinfopakistan.ucoz.com/
Lysosomal enzymes synthesized in endoplasmic reticulum are marked with specific mannose-6 phosphate as marker molecules; they are then bind to their specific receptors, then they pinched off as primary vacuoles using clathrin proteins as a coat, which fuses with primary lysosomes or phagosomes. The mannose receptors are recycled into trans-golgi membranes.
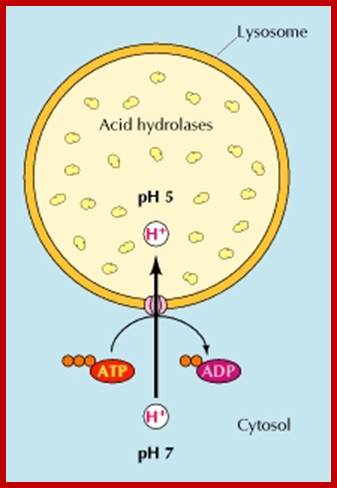
Regular pumping of protons into across Lysosomal unit membrane into Lysosomal sap (pH4.2) maintain their Lysosomal enzyme in dormant condition; http://www.ncbi.nlm.nih.gov/
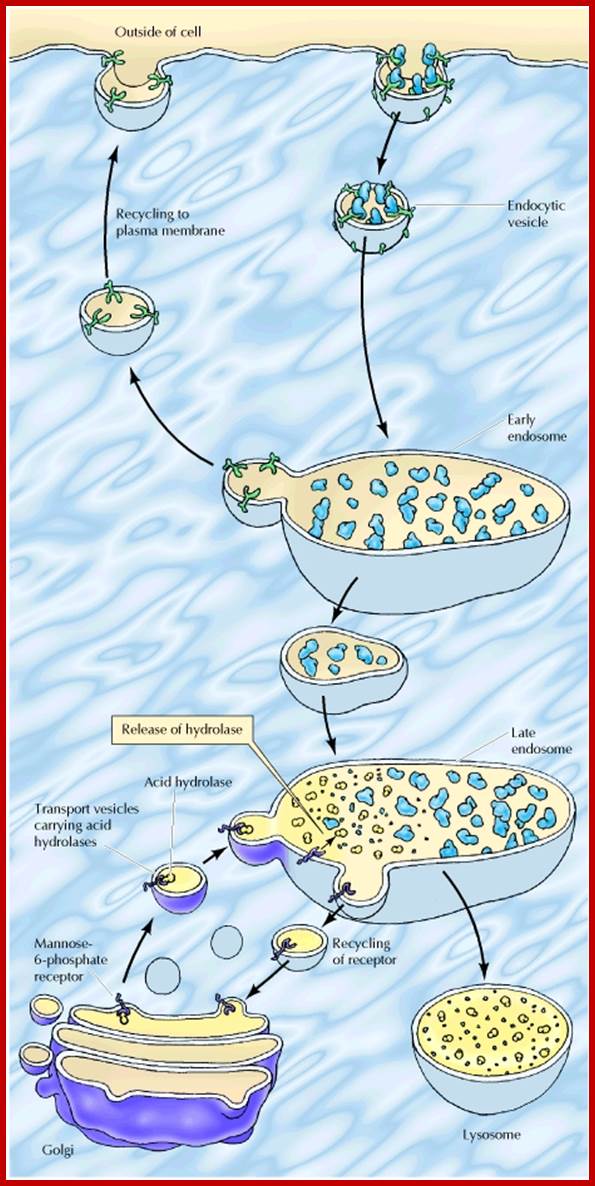
http://www.ncbi.nlm.nih.gov/p; http://cc.scu.edu.cn/
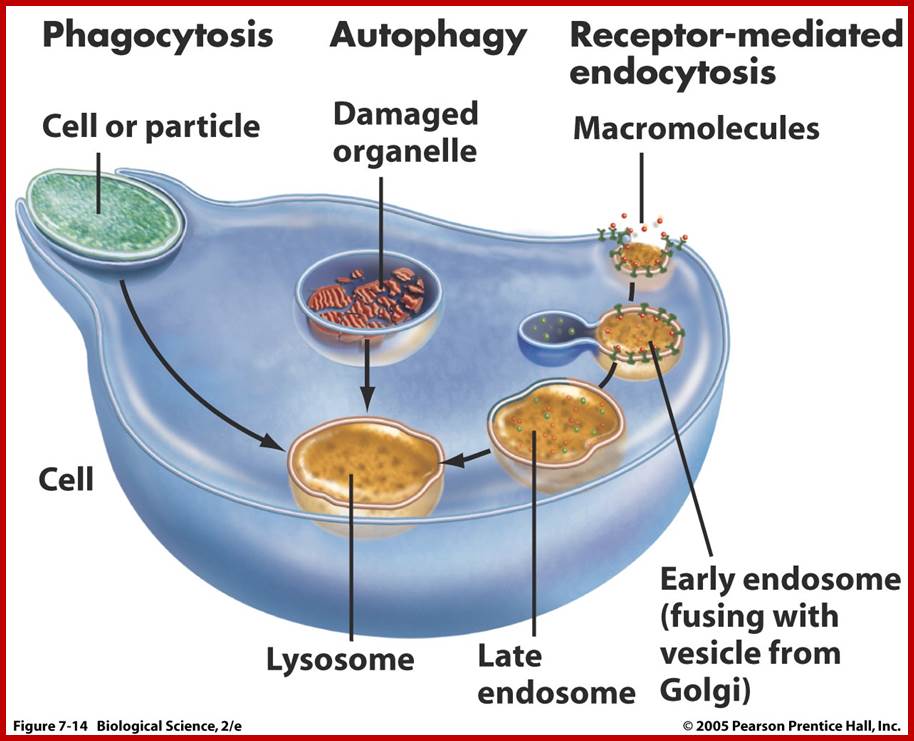
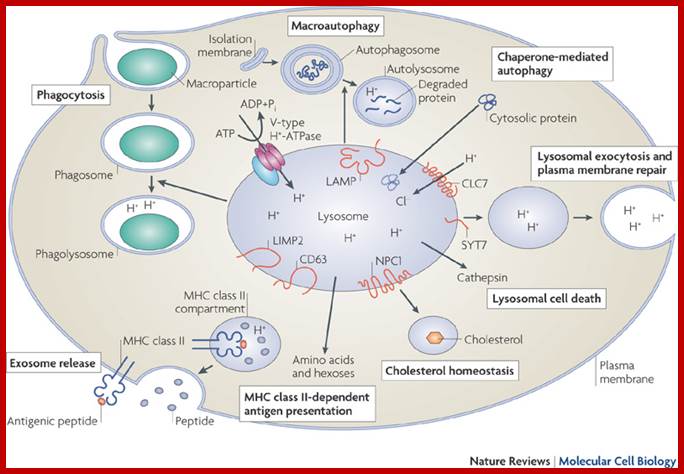
Lysosomes are important organelles in degrading many components. Lysosomes are important in maturation of phagosomes to phago-lysosomes; then to lysosomes.
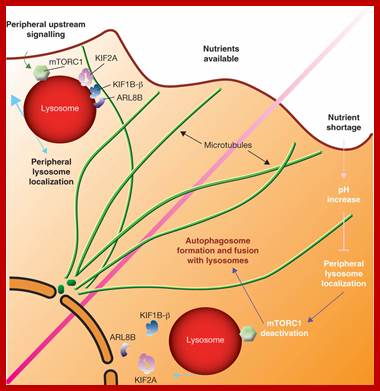
Under conditions of nutrient availability, lysosomes are maintained at the periphery of the cell through a microtubule-dependent mechanism involving kinesins (KIF1B-β and KIF2A) and the monomeric GTPase ARL8B. mTORC1, which is bound to the cytoplasmic surface of lysosomes, is activated by upstream signalling when at the cell periphery. In the absence of nutrients, the increase in cytoplasmic pH prevents the transfer of lysosomes to a peripheral location through the release of kinesins and ARL8B from microtubules, resulting in the accumulation of lysosomes in the perinuclear area and inactivation of mTORC1. A major consequence of mTORC1 inactivation is the stimulation of autophagosome formation and the fusion of autophagosomes with lysosomes in the perinuclear area. http://www.nature.com/
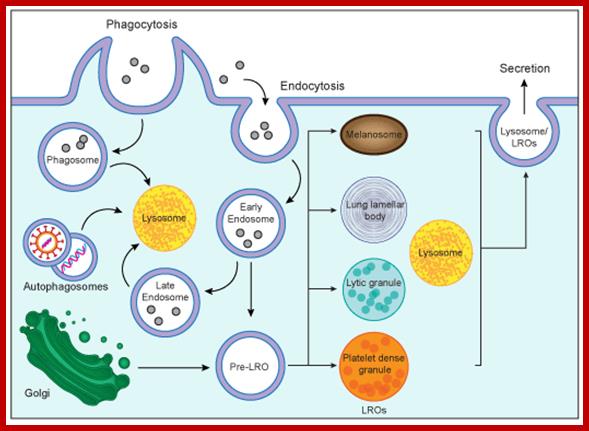
Biogenesis of lysosomes and lysosome-related organelles (LORs); Lysosomes are cytoplasmic organelles responsible for degradation within a cell. LROs share some features with lysosomes but have distinct morphologies and functions. https://mynotebook.labarchives.com
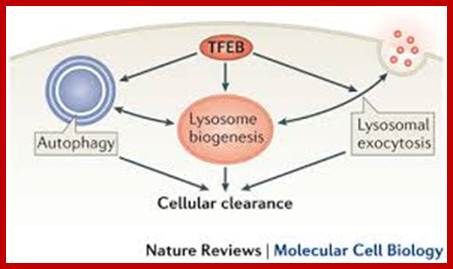
Transcription factor EB (TFEB) controls lysosomal biogenesis by regulating the level of lysosomal enzymes, lysosomal acidification and the number of lysosomes. TFEB also controls autophagy by regulating the number of autophagosomes and the fusion between autophagosomes and lysosomes. Finally, TFEB regulates the docking and fusion of lysosomes to the plasma membrane in the process of lysosomal exocytosis. The concerted action of these three processes leads to cellular clearance. Carmine Settembre, Alessandro Fraldi, Diego L. Medina & Andrea Ballabio;www.nature.com

Biogenesis of lysosomes; www.circresearch.com
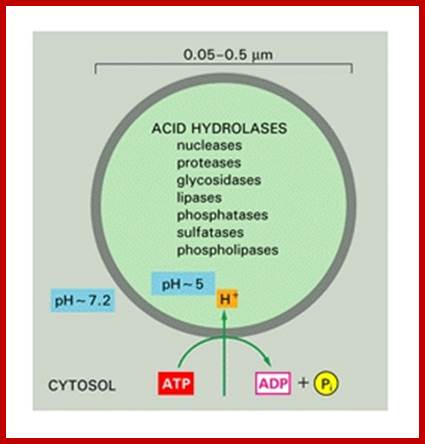
Some acid hydrolases; http://ztopics.com/
http://www.youtube.com/
https://igm.jhmi.edu
Plant Cell central Vacuoles;
They act like animal lysosomes. They can breakdown large molecules under acidic conditions for the vacuole sap is acidic. Plant central vacuole develops from smaller provacuolar structures and Golgi vesicles. As plant cell expands the central vacuole increase in size and occupies more than 90% of the cell space and the all cell organelles pushed to peripheral region. Central vacuole is formed due to of multiple membrane vesicles. In Arabidopsis, TIP3;1 and TIP2;1 are likely to use a Golgi-independent pathway, while TIP1;1 is likely to use a Golgi-dependent pathway. Tonoplast has a regulatory role in movement of ions. The Vacuole acts as storage organ, also stores toxic components, it provides pH and ionic homeostasis; it also acts a defense organelle.
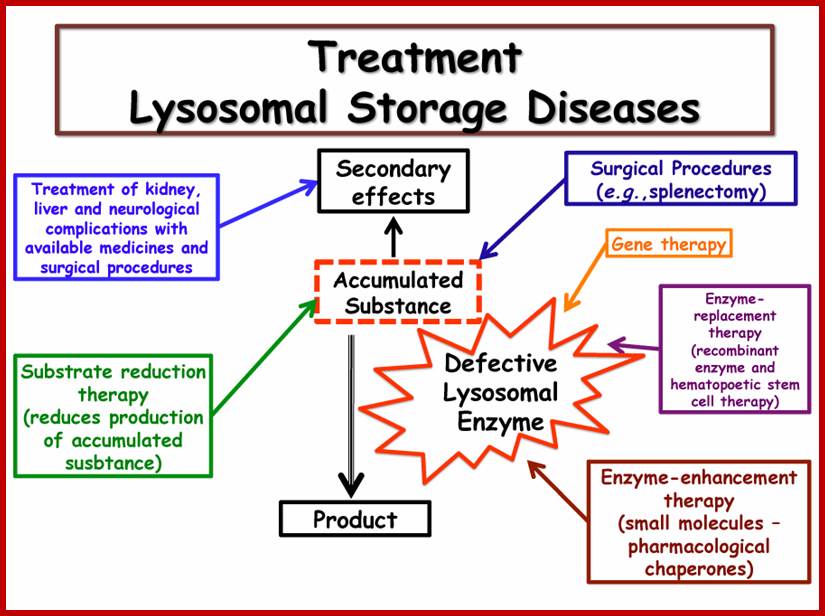
Lysosomal diseases:
Storage disorders, lack of specific lysozymal enzymes can create problems, it can lead organ disorders, cause disease called Gaucher’s disease (lack lipids) and Tay-Sachs disease and Pompe's disease- lack of glucocerebrosidase, several neurodegenerative disorders, cancer, cardiovascular diseases, and ageing-related diseases; at least 30 genetic diseases.