CHROMOSOMES;
They are fine threads made up of DNA and proteins compacted into genetically strong threads called chromosomes, for they give color when stained acetocarmine or fuchsin. They are localized in nucleus in higher organisms, but suspended in bacteria as DNA protein threads. In viruses the genetic material can be RNA or DNA are compacted in proteinaceous capsids.
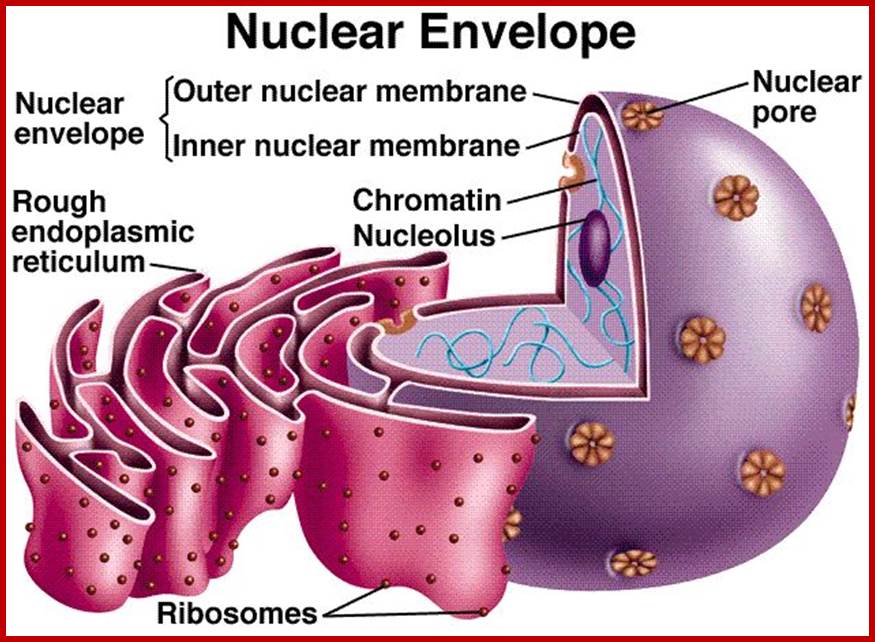
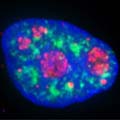
Nucleus with extended nuclear membrane (with nuclear pore complex-look like stars) into ER, inside the envelope one finds nucleolus and chromosome; www.mthira.vic.edu.au
Number: The number of chromosomes varies from species to species, but it is constant and characteristic to a given species. Generally the number is denoted by terms like Haploid (n) and Diploid (2n) etc. The haploid consists of one set of chromosomes, where every chromosome is unique in its structure, morphology and genetic content and form one set of chromosomes. The diploid consists of two such haploid sets of chromosomes. Ex: Homo sapiens 2n=46, Pisum sativum 2n=14, Phaseolus vulgaris 2n=14, Allium cepa 2n=16, Ascaris megalocephala univalens 2n=2. Polyploid indicates the number of haploid sets; any thing more than two, like Triploid (3n), Tetraploid (4n) etc. highest number of chromosomes are found in Ophioglossum a primitive fern 1260- 630 pairs..
Common species- only few-
|
|

|
C Common name |
Specific name |
Chromosomal number(2n) |
Fruity fly |
Drosophila |
8 |
|
Frog |
Rana pipiens |
26 |
|
Gorilla |
Gorilla gorilla |
48 |
|
Monkey |
Macaca mulatta |
42 |
|
Man |
Homo sapiens |
46 |
|
Garden pea |
Pisum sativum |
14 |
|
French bean |
Phaseolus vulgaris |
14 |
|
Onion |
Allium cepa |
16 |
|
Cabbage |
Brassica oleracea |
18 |
|
Coffee |
Coffea Arabica |
44 |
|
Potato |
Solanum tuberosum |
24 |
|
Fern Ophioglossum |
Ophioglossum reticulum/vulgatum
|
1260?
|
|
Chimpanzee pan trolodytes |
Two chromosomes 2a and 2b fused at telomere ends and produced Hu chromo 2; 2n=46 Inversions in 1,4,5,9,12,15,16,17 and 18 |
48 |

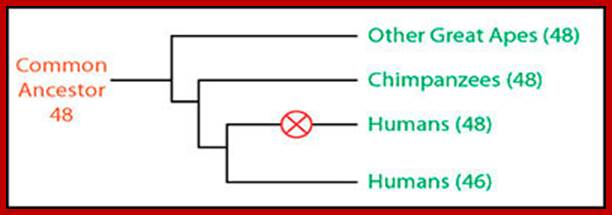
Human Ancestors; www.apologeticspress.org

Chimpanzees are humans' closest relatives. They are native to central and west Africa; Human with 2n=46 chromosomes and chimps 2n=48 chromosomes; Chimps 2A and 2B chromosomes, a part of 9 have fused to generate 2n=46; http://www.dailymail.co.uk/
 [Chimpanzees] make tools, use language, understand symbols
and build shelters. They also develop long-term bonds, live in highly social
groups, make jokes, manipulate, deceive, empathize, and show care for other
members of their group and other species. The behavioral differences have been
relegated to artificial human-constructed continuums of complexity.
[Chimpanzees] make tools, use language, understand symbols
and build shelters. They also develop long-term bonds, live in highly social
groups, make jokes, manipulate, deceive, empathize, and show care for other
members of their group and other species. The behavioral differences have been
relegated to artificial human-constructed continuums of complexity.
;http://theadvancedapes.com/

Apes; Bonobos

https://answersingenesis.org
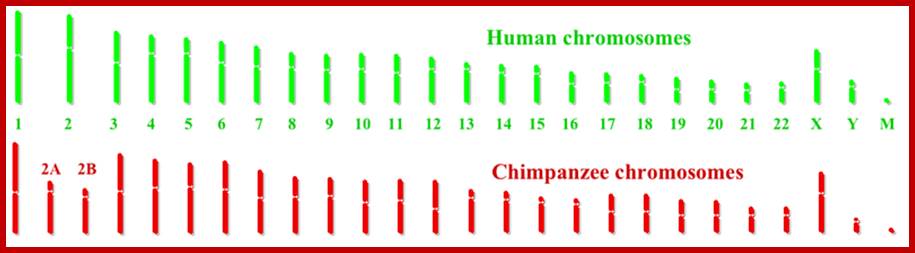
Human and Chimpanzee genomes and M stands for Mitochondrial DNA, modern human 6.5 billion, mitochondrial DNA all over the world has been inherited from few south African mothers; http://en.wikipedia.org/
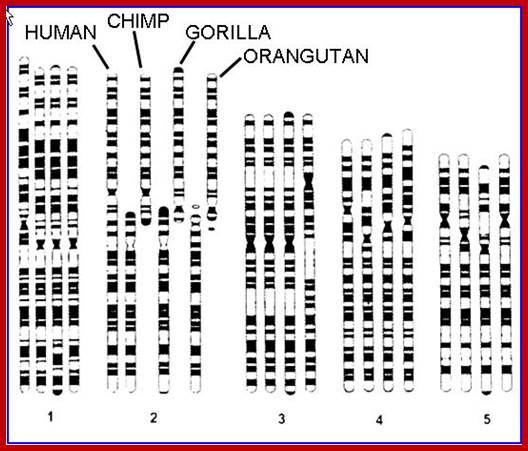
Humans have 46 chromosomes, whereas chimpanzee, gorilla, and orangutan have 48. Human chromosome 2 was formed by the head-to-head fusion of two ancestral chromosomes that remained separate in other primates. Sequences that once resided near the ends of the ancestral chromosomes are now interstitially located in 2q13–2q14.1. Portions of these sequences had duplicated to other locations prior to the fusion. The “Amelogenin”’ gene the female sex linked gene? https://www.ncbi.nlm.nih.gov/
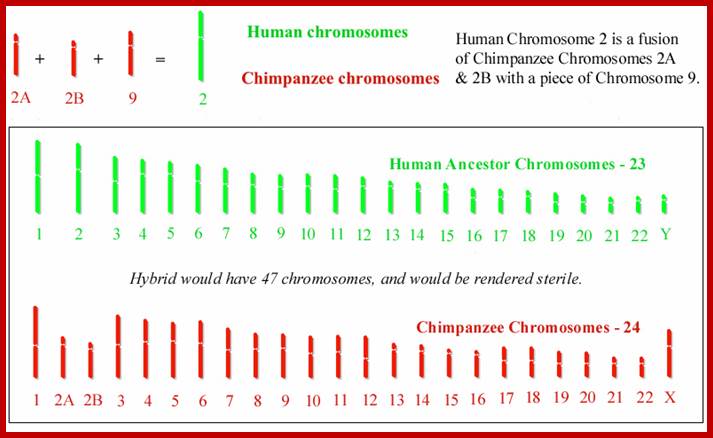
http://originalthinkingman.tripod.com/

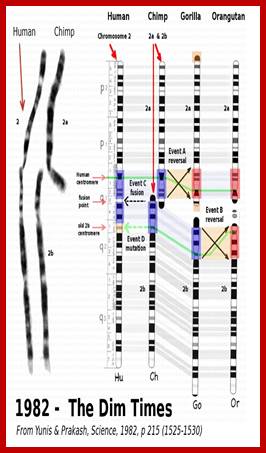
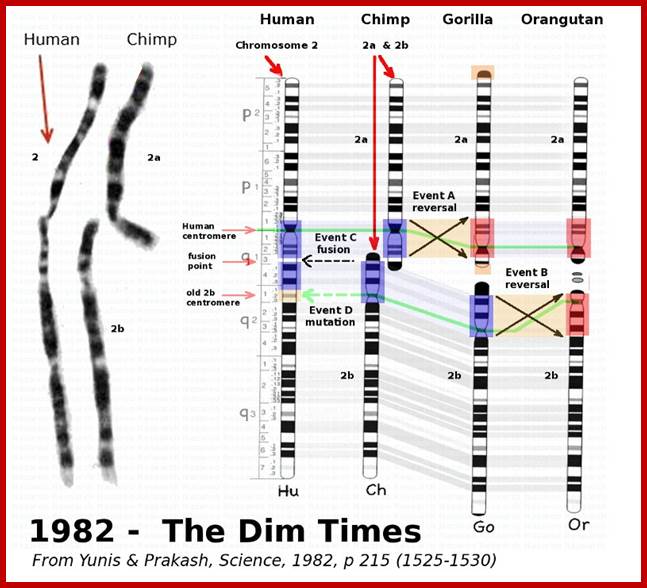
Depiction of a hypothetical scenario in which chimpanzee chromosomes 2A and 2B fused end-to-end to form human chromosome 2. All chromosomes were comparatively drawn to scale according to cytogenetic images by Yunis and Prakash (1982). Putative fusion and cryptic centromere sites were placed on human chromosome 2 based on current locations in the UCSC Genome Browser. Note the extreme lack of positional correspondence for the cryptic centromere site and to a lesser extent, the fusion site and the current human centromere. Also note the size discrepancy which is about 10% or 24 million bases.
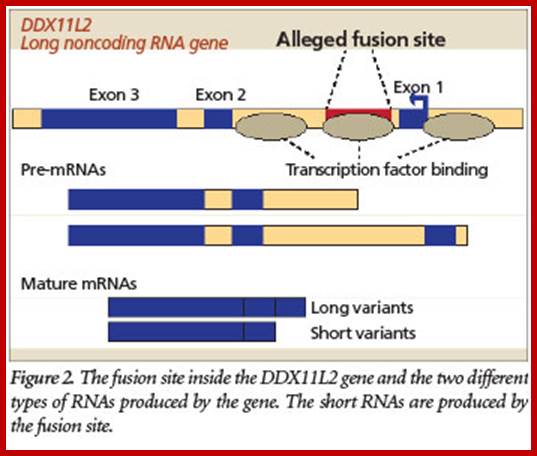
Alleged Human Chromosome 2 “Fusion Site” Encodes an Active DNA Binding Domain Inside a Complex and Highly Expressed Gene—Negating Fusion; by Jeffrey Tomkins on October 16, 2013; https://answersingenesis.org;
The purported fusion site is located inside not as shown in the above figure. More specifically, a crucial RNA helicase gene called DDX11L2 that produces long non-coding RNAs. This gene is expressed in at least 255 different tissue and cell types in humans and is highly coregulated with many other important genes in the cell. So not only is the gene highly active throughout the human body, it is tightly networked with many other genes, including those that are involved in the development of blood cells.
http://www.icr.org/
But the evolutionary fusion story gets worse. The fusion-like sequence itself has an important functional purpose based on recent data available at the UCSC Genome Browser (genome.ucsc.edu) genomic database. Specifically, the fusion site sequence binds to at least 11 different transcription factors, including RNA polymerase II, the key enzyme that transcribes genes. Transcription factors are specialized proteins that turn genes off and on. The fact that these proteins specifically bind to the alleged fusion site sequence indicates that it is a promoter located inside the gene (Figure). It is common for human genes to have these promoter regions located both in front of the main body of the gene and inside them.
The overwhelming lack of evidence for chromosome 2 fusion does not support the evolutionary story in any feasible way. Clearly, the so-called fusion sequence is an important functional feature called a promoter inside a highly expressed gene, not an accident of evolution. Research shows that Chimps 2a and 2b does not fuse end to end, but the fusion site is located elsewhere, however theory of fusion of 2a and 2b not excluded. However axiomatic evidence show human evolved from shared ancestor with Chimpanzee and Bonobos. Fusion provides evidence from banding pattern- 2a and 2b are fused end to end, among two centromeres one is functional another inactive and the internal telomere sequence within human chromosome 2. All other chromosomal maps are similar to chimps (Pan troglodytes). Another possibility is the human ancestors themselves created chromosome 2 by their 2a ans 2b chromosomal fusion; http://www.icr.org/
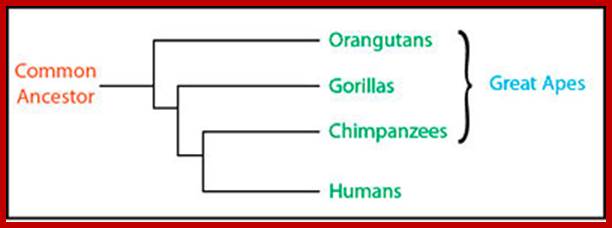
Chimpanzee and bonobo or pigmy chimps are grouped together. http://apologeticspress.org/
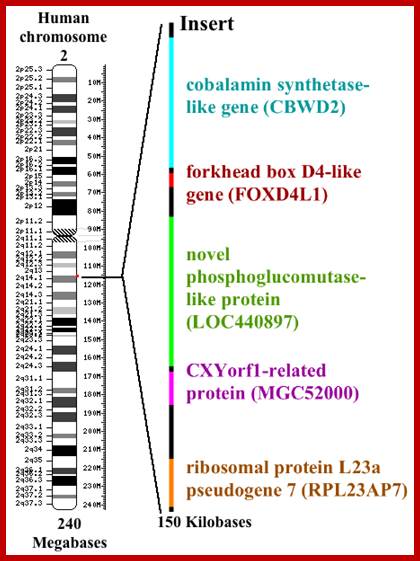
Chromosome 2 insert.png; I made this diagram using Phtsjok45.wordpress.com, Oto Shop and Claris Draw. J.W Schmidt;
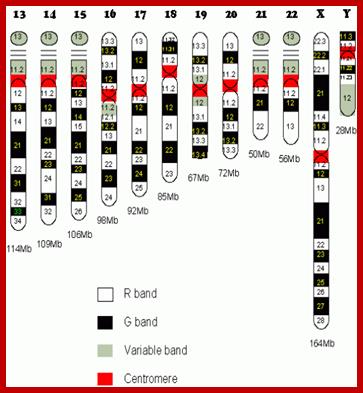
Human chromosome karyotype; look at the secondary constriction on chromosomes13,14,15,21 and 22; Chromosome 2 is believed to be originated from chimpanzee chromosomes 2A and 2B; Analysis further showed 7600 genes shared between two genomes; certain Genes found as forkhead-boxP2 transcription factor which is involved in human speech acquisition and hearing ability. www.thelancet.com.
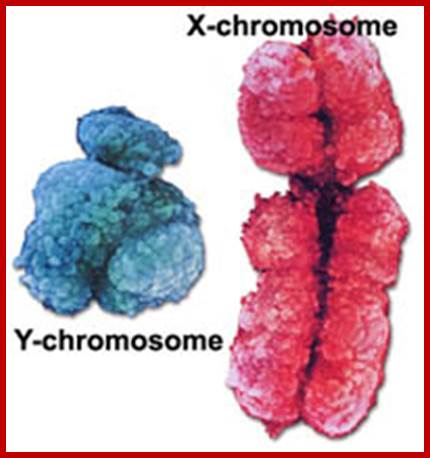
Human; www.musc.edu
Basic structural feature of a chromosome-P arm (shorter) and Q arms (longer) and centromere and positions of genes in p and q of a chromosomes; www. galleryhip.com
If there are any changes from the normal diploid number, like loss or addition of one or two chromosomes leads to Aneuploidy. Polyploidy and aneuploidy are the variations from the normal diploid number. Such changes often produce variations in the morphology and functions of plants and animals.
www.koanicsoul.com; http://bhavanajagat.com/
https://www.pinterest.com
Modern humans (Homininis) are 97% similar to chimps, but have one fewer chromosome. Our makers fused two of the ape chromosomes together, so 24 became 23. The reason you would do this is to make ape DNA compatible with alien DNA, which presumably had 23 chromosomes. Humans also have 9 “inversions”. An inversion is a segment of ape DNA that’s been flipped and reinserted. All humans have these, which is incredibly improbable (because inversions are recessive and non-carriers had to all die out after each one sequentially occurred by chance). This supports the genetic engineering theory. Within our 20,000 to 25,000 genes (3,234.83 Mb) there are 223 that have no analogues in any other animal species, although 113 of the 223 can be found among bacteria. Thus, mainstream science concludes that all 223 of the “unique” human genes have somehow been put into our genome by “lateral transfer,” which means infection by bacteria. But what about the other 110 genes that aren’t found any other systems, are they unique to human race? Most human Y chromosome sequences thus far examined do not have homologues [same relative position or structure] on the Y chromosomes of other primates. Speculation about the disappearance of Y’ chromosome some time later; it can be few thousand yrs or ten thousand years or more? Human female X chromosomes do look somewhat ape like, but not the male’s Y. This means that if humans are a crossbred species, the cross had to be between a female ape-like creature (i.e, “creature of Earth”) and a male being from elsewhere. Also, by certain methods of DNA dating, one can tell that numerous genes have been recently added to the human genome.
Man, and Apes exist with genomes that are almost identical and yet man cannot directly descend from the anthropoid apes. The fusion of Chromosomes #2 a, and #2 b would cause the production of a non-viable mutant or an individual who may not produce any offspring. Firstly, we need a male and a female with this fused chromosome in their gametes; the sperm and the egg to produce an offspring with a unique set of 46 chromosomes; 22 pairs of autosomes and a pair of (XY or XX) of Sex Chromosomes X and Y. It requires two identical, rare mutants, male and female to produce offspring and to establish an entirely new population. The Theory of Evolution proposes that random, unguided, mutations lead to changes in a Species and eventually lead to its descent as a new Species. If fusion of two, distinct, separate chromosomes such as #2 a, and #2 b is required, the fusion event must happen in a Hominid population about 10,000 years ago and this population would have no relationship with the living apes (Bhavanajagut)
.
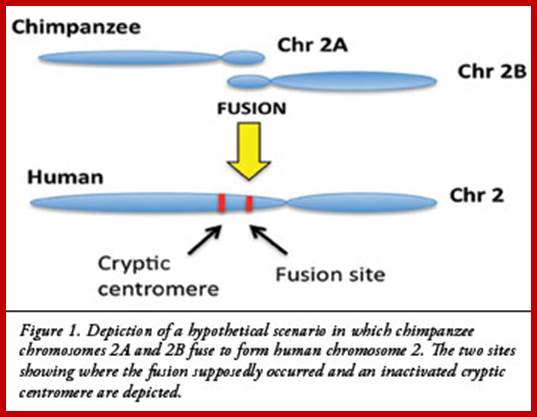
According to the evolutionary interpretation, this fusion took place at some point after humans and great apes split into different evolutionary branches. Figure 1 presents a pictorial depiction of this hypothetical event. A majority of the data for the fusion model is based on indirect laboratory techniques that were commonly used prior to the revolution in genomics, before DNA sequencing became routine and cost-effective. A key article published in 2002 actually analyzed the DNA sequence evidence at the key chromosomal fusion sites in humans and produced more questions than answers. This author and Dr. Jerry Bergman, a professor at Northwest State College in Ohio, recently completed a new analysis of the scientific literature and available DNA sequence data that seriously calls into question the validity of the fusion model and human evolution in general. Our research on the human chromosome 2 is tentatively scheduled to be published in theJournal of Creation. A brief summary of the major points discovered in this exciting new chromosome 2 research will be outlined in an upcoming issue of Acts & Facts.;Jeffrey P.Tomkins, PhD; http://www.icr.org/

http://blogs.discovermagazine.com/A copy of StSat got glued to the end of the red chromosome, and then the pink and green segments at the top of the green chromosome got flipped. The chromosomes at the right of the figure show you what our two chromosomes looked like before they got fused. When the human and chimpanzee lineages split, each lineage inherited them. And in each lineage, they evolved in a different way. In the chimpanzee lineage, the chromosomes didn’t fuse. Instead, this happened:
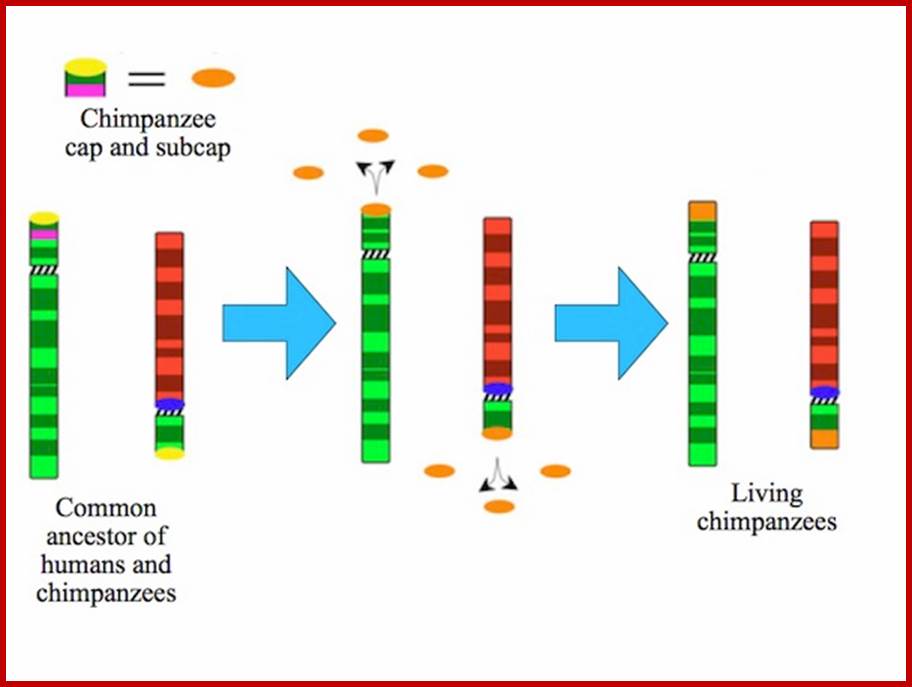
http://blogs.discovermagazine.com/ The caps on both the green and red chromosomes were duplicated massively and ended up on lots of other chromosomes. And finally, here’s what happened to humans after our ancestors split from chimpanzees:
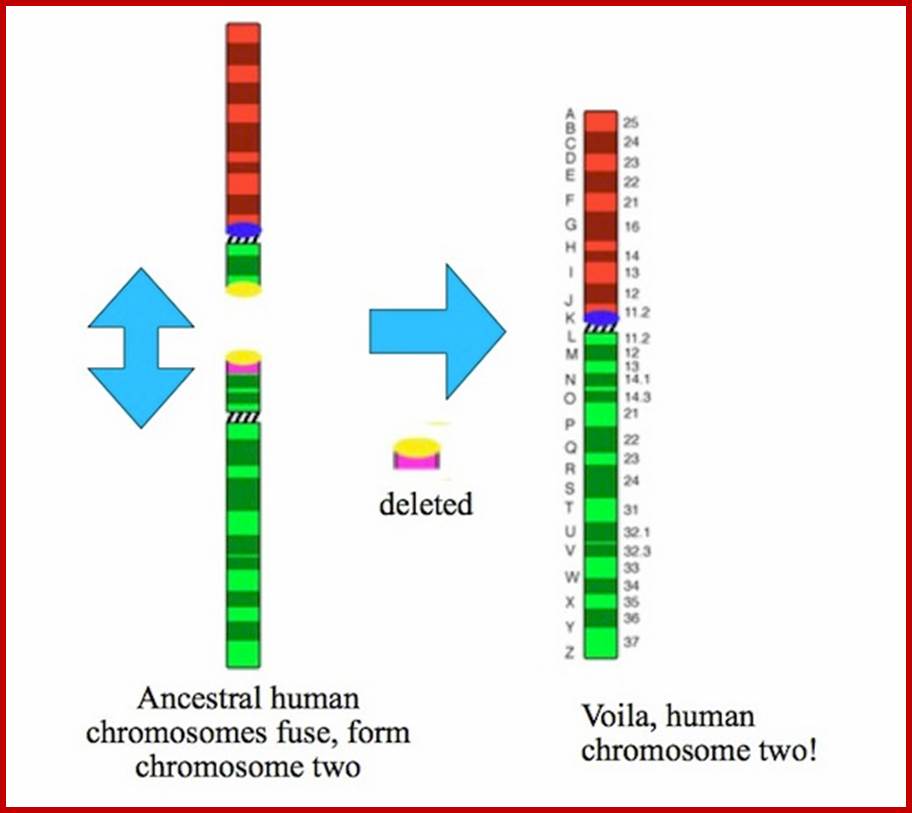
The two chromosomes fused, and the cap was deleted, including St Sat. It could no longer spread around our genome, the way it did in chimpanzees and gorillas. This study is an important advance in our understanding of how human chromosomes evolved–a subject of medical significance, too, since the duplication of the DNA at the end of chromosomes can cause dangerous mutations that can cause genetic disorders. Plus, it is very cool to see how our chromosomes are, in fact, an ancient patchwork. http://blogs.discovermagazine.com/

Ideogram of human chromosome 2. Mbp means mega base pair. See locus for other notation.

Evidence for the fused 2a and 2b chromosome comes from Telomeric sequence and fused chromosomal sequence. From this image above of several sequences of human DNA, you can see that the telomere at the end of the chromosome has the sequence TTAGGG repeated many times, and although the repeated sequences are not uniform in the fused telomere sequence (because of other point mutations which have taken place since the fusion event), nevertheless, right in the middle of the chromosome, you can clearly see TTAGGG repeated (imperfectly) many times followed closely by a repetition of the opposite sequence CCCTAA. We can therefore see the exact point where the two chromosomes fused. Mystery solved! ; chromosome 2 is consistent with the common ancestry of humans and other apes. According to researcher J. W. IJdo, "We conclude that the locus cloned in Cosmids c8.1 and c29B is the relic of an ancient telomere-telomere fusion and marks the point at which two ancestral ape chromosomes fused to give rise to human chromosome 2; It is likely 2a and 2b chromosomes of ancestors of humans might have broken and fused by an unknown mechanism-https://en.wikipedia.org/wiki
researcher J. W. IJdo, https://aperimentis.wordpress.com/2015
It is also possible that humans lived many thousands of years before the present homo sapiens, contained 48 chromosomes. During a period of time, 2a and 2b fused by broken chromosomes fused telomere to telomere. Gene analysis tells the same story as found in the case of ‘Ape’s’ chromosomes. To live and adjust genetically it is easy than the chromosomal fusion of apes and transformation. Chromosomal fusion does not provide any genetic clue for evolving humans for there is change in genetic composition of human like humans to present humans.

(Data source: Ensembled genome browser release 68, July 2012)
https://en.wikipedia.org
Human chromosome numbers, size of the genome and genes:
Chromosome Number and DNA size 106 bp
1. 249. 249 mbp (247)
2. 243. 243 mbp (242
3. 1445. 198 mbp (199
4. 1023. 191 mbp
5. 1261. 180 mbp
6. 1401. 171 mbp
7. 1086. 159 mbp
8. 1042. 146 mbp
9. 1626. 141 mbp
10. 477. 135 mbp
11. 821. 135 mbp
12. 915. 133 mbp
13. 1139. 115 mbp
14. 1471. 107 mbp
15. 408. 102 mbp
16. 1715. 90 mbp
17. 762. 81 mbp
18. 357. 78 mbp
19. 657. 59 mbp
20. 63 63 mbp
21. 48 48 mbp
X. 1090 (800). 155 mbp
Y. 144 (50). 59 (4) mbp
Note 1pg = 978bp; convert base pairs into pictograms; Number of base pairs = mass in pg X 0.978 x 10^9; 1kb = 10^3; 1Mb = 10^6bp, and 1Gb = 10^9bp; Mt DNA-16mbp;
Present status; human contain ~21,000 genes; total human DNA in BP= 3,079,843,747
Human reference genome, with links to Data source: Ensembl genome browser release 68, July 2012).
|
Chromosome |
Total base pairs |
% of bases |
Sequenced base pairs[26] |
|
|
2000 |
247,199,719 |
8.0 |
224,999,719 |
|
|
1300 |
242,751,149 |
7.9 |
237,712,649 |
|
|
1000 |
199,446,827 |
6.5 |
194,704,827 |
|
|
1000 |
191,263,063 |
6.2 |
187,297,063 |
|
|
900 |
180,837,866 |
5.9 |
177,702,766 |
|
|
1000 |
170,896,993 |
5.5 |
167,273,993 |
|
|
900 |
158,821,424 |
5.2 |
154,952,424 |
|
|
700 |
146,274,826 |
4.7 |
142,612,826 |
|
|
800 |
140,442,298 |
4.6 |
120,312,298 |
|
|
700 |
135,374,737 |
4.4 |
131,624,737 |
|
|
1300 |
134,452,384 |
4.4 |
131,130,853 |
|
|
1100 |
132,289,534 |
4.3 |
130,303,534 |
|
|
300 |
114,127,980 |
3.7 |
95,559,980 |
|
|
800 |
106,360,585 |
3.5 |
88,290,585 |
|
|
600 |
100,338,915 |
3.3 |
81,341,915 |
|
|
800 |
88,822,254 |
2.9 |
78,884,754 |
|
|
1200 |
78,654,742 |
2.6 |
77,800,220 |
|
|
200 |
76,117,153 |
2.5 |
74,656,155 |
|
|
1500 |
63,806,651 |
2.1 |
55,785,651 |
|
|
500 |
62,435,965 |
2.0 |
59,505,254 |
|
|
200 |
46,944,323 |
1.5 |
34,171,998 |
|
|
500 |
49,528,953 |
1.6 |
34,893,953 |
|
|
800 |
154,913,754 |
5.0 |
151,058,754 |
|
|
50 |
57,741,652 |
1.9 |
25,121,652 |
|
|
Total |
21,000 |
3,079,843,747 |
100.0 |
2,857,698,560 |
Table (above) summarizes the physical organization and gene content of the human reference genome, with links to the original analysis, as published in the Ensemble database at the European Bioinformatics Institute (EBI) and Welcome Trust Sanger Institute. Chromosome lengths were estimated by multiplying the number of base pairs by 0.34 nanometers, the distance between base pairs in the DNA double helix. The number of proteins is based on the number of initial precursor mRNA transcripts, and does not include products of alternative pre-mRNA splicing, or modifications to protein structure that occur after translation.
The number of variations is a summary of unique DNA sequence changes that have been identified within the sequences analyzed by Ensemble as of July, 2012; that number is expected to increase as further personal genomes are sequenced and examined. In addition to the gene content shown in this table, a large number of non-expressed functional sequences have been identified throughout the human genome (see below). Links open windows to the reference chromosome sequence in the EBI genome browser. The table also describes prevalence of genes encoding structural RNAs in the genome.
Info about Mitochondria;
"Eve" with mitochondrial DNA, lived in Africa about 150,000 years ago. Other scientists studying the male Y chromosome have reached similar conclusions. The new Herto fossils are from a population living at exactly this time. Like mitochondrial "Eve", Y-chromosomal "Adam" probably lived in Africa. A recent study (March 2013) concluded however that "Eve" lived much later than "Adam" – some 140,000 years later. (Earlier studies considered, conversely, that "Eve" lived earlier than "Adam".) More recent studies indicate that mitochondrial Eve and Y-chromosomal Adam may indeed have lived around the same time. Mitochondrial Eve is the most recent common matrilineal ancestor, not the most recent common ancestor.
Mitochondrial Eve is estimated to have lived between 99,000 and 200,000 years ago, most likely in East Africa, when Homo sapiens sapiens (anatomically modern humans) were developing as a population distinct from other human sub-species.
Mitochondrial Eve lived later than Homo heidelbergensis and the emergence of Homo neanderthalensis, but earlier than the out of Africa migration. The dating for "Eve" was a blow to the multiregional hypothesis and a boost to the theory of the origin and dispersion of modern humans from Africa, replacing more "archaic" human populations such as Neanderthals. As a result, a consensus emerged among anthropologists that the latter theory was more plausible.
Analogous to the Mitochondrial Eve is the Y-chromosomal Adam, the member of Homo sapiens sapiens from whom all living humans are descended ‘patrilineally’. The inherited DNA in the male case is his nuclear Y chromosome rather than the mtDNA. Mitochondrial Eve and Y-chromosomal Adam need not have lived at the same time. For example, Y-chromosomal Adam has been estimated to have lived during a wide range of times from 180,000 to 581,000 years ago, while a 2013 paper concluded that he lived between 120,000 and 156,000 years ago (however, this paper did not include some Cameroonians and one African American, who did not inherit their Y from that "Adam"[
Mitochondrial DNA (mtDNA) and Y-chromosome DNA are commonly used to trace ancestry in this manner. mtDNA is generally passed un-mixed from mothers to children of both sexes, along the maternal line or ‘matrilineally’. Human epigenome
· Mitochondrial Eve, the most recent female-line common ancestor of all living people.
· "Y-chromosomal Adam", the most recent male-line common ancestor of all living people

In human genetics, Mitochondrial Eve is the matrilineal most recent common ancestor (MRCA) of all currently living humans. This is the most recent woman from whom all living humans today descend, in an unbroken line, on their mother’s side, and through the mothers of those mothers, and so on, back until all lines converge on one woman, who is estimated to have lived approximately 100,000–200,000 years ago. Because all mitochondrial DNA (mtDNA) generally (but see paternal mtDNA transmission) is passed from mother to offspring without recombination, all mtDNA in every living person is directly descended from hers by definition, differing only by the mutations that over generations have occurred in the germ cell mtDNA since the conception of the original "Mitochondrial Eve".https://en.wikipedia.org
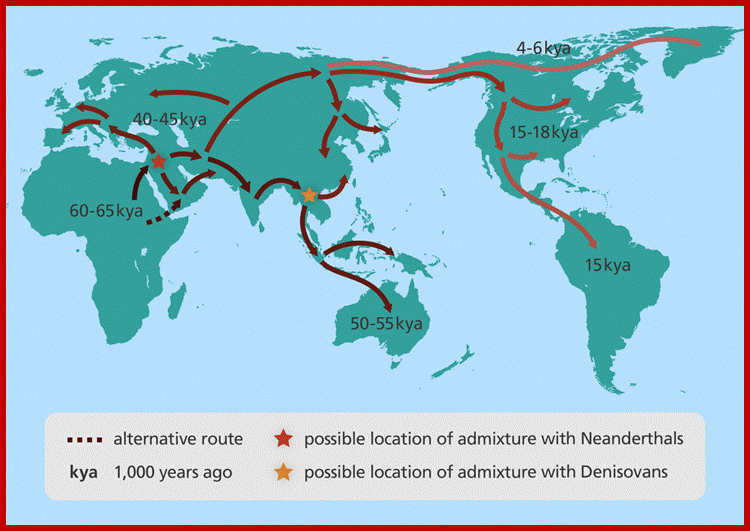
A map showing human migration out of Africa. Image credit: Genome Research Limited
On the biggest steps in early human evolution scientists are in agreement. The first human ancestors appeared between five million and seven million years ago, probably when some apelike creatures in Africa began to walk habitually on two legs. 160,000 yrs old human anatomy was discovered in Ethoipia. Neanderthals and the recently discovered Denisovans of Siberia were two of several extinct types of humans with whom we lived side by side, Neanderthals disappeared from this earth around 30,000 yrs ago.
Tree of humans and apes; https://www.mpg.de

https://med.stanford.edu/
Many species that were more closely-related to humans have lived and become extinct since the time of the chimpanzee- human ancestor. They are collectively called Hominins. One of the Hominin is the Neanderthal, whose lineage diverged from ours 300,000–500,000 years ago. Neanderthals lived in western Eurasia, sometimes alongside our ancestors, until they became extinct around 30,000 years ago. Max-Plank Gesellschaft; https://www.mpg.de/

96% of chimp DNA is same in Humans, with some 2A and 2b and small chip of chromosome 9 are fused to develop into human species; only forty million base pairs differ between human and chimp.; http://www.philipcaruso-story.com/

Human genes categorized by function of the transcribed proteins, given both as number of encoding genes and percentage of all genes.
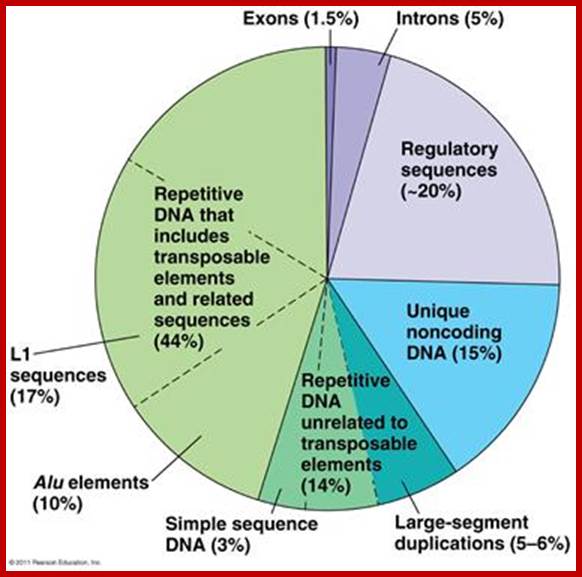
Human Genome; www.theologyonline.com
All "male benefit" spermatogenic genes on the Y have accumulated in ampliconic regions of repeat DNA sequences and inverted repeat DNA sequences. Originally the Y-chromsome has 1438 gene, and now they retain only 60 genes (9 family).Y chromosome and male infertility; Experts predicts that male determining Y chromosomes get lost in another five million years for the depiction is due to the finding the loss of genes from Y chromosomes have already started; If what happens to males and reproduction; No! It is impossible; one cannot assume the human to be only female, how do they reproduce? If it comes true, human race will be extinguisfed? www.infertile.com
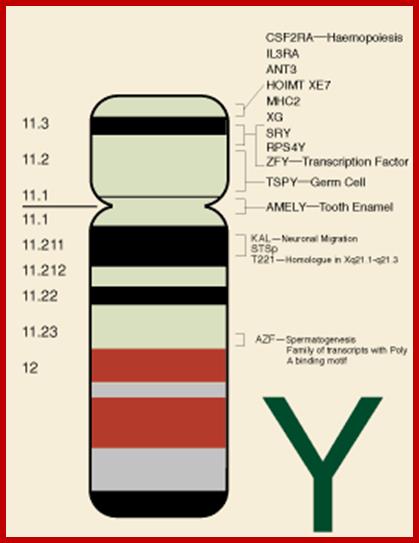
Some known genes in Y-chromosome.www.ucl.ac.uk
Unique exons-1.5%, Introns-5%, regulatory sequences-~20%, Unique noncoding sequnces-15%, Repetitive DNA (excluding. Transposable elements), Repetitive sequences includes transposable elements and its related sequences (L1 seuqnces-17%,), Alu elements-10%, simple sequences-3%and Large segment duplicated sequences-5-6%
Size and shape: The size of chromosomes varies from as small as 0.1um to as big as 15 um in length. But the size remains constant for any given chromosome of a given species.
The shape of chromosome depends upon the position of the centromere; it is a misnomer. During anaphasic movement depending upon the position of the centromere they show shape, otherwise they are rod shaped. Still text books it is loosely used in describing the shape. Accordingly, if the centromere is in the middle of a chromosome (metacentric) the chromosome assumes “V” shape. Chromosomes having sub-metacentromere show up as "J" shape.
Acrocentric (centromere located just behind telocentromere of the chromosome) and telocentric (centromere chromosomes present at the extreme end), look like rod shaped chromosomes during anaphasic movements. But the Diffused or Holocentromeric (centromeric activity found all along the length of the chromosome) and polycentromeric (centromere present in two or more loci) chromosomes look like horizontal rod shaped or wavy shaped respectively. Acentric are those chromosomes which lack in centromeres. They move randomly and often get lost during mitotic and meiotic divisions.
Viral particles and prokaryotic bacteria contain their genetic material either packed into their capsids or suspended in the cytoplasm respectively. Association of proteins gives stability to DNA molecules. Bacterial DNA is bound to histone like such as Hn, H, GLP and P which show greater similarities to histone proteins. SV 40 viral DNA molecules, after infection get associated with regular eukaryotic chromosomal proteins and behave like mini chromosomes. However, eukaryotes contain the genetic material, which is highly organized into complex structural units called chromosomes. But in euglena, the chromosomes show a simplified packaging of DNA fibrils, where the DNA is transversely packed. Such chromosomal structures truly reflect the transitional organization from prokaryotic bacteria to eukaryotic chromosomes.
GROSS STRUCTURE:
Structure;
Chromosomes are often called as the vehicles of heredity or physical bridge between two generations or as the physical basis of inheritance. The credit for the discovery and naming of chromosomes should go to Brown and Waldeyer.
Chromosomes show three levels of structural organization, such as primary, secondary and tertiary. Chromosomes, which appear under light microscopical level, show tertiary level of organization.
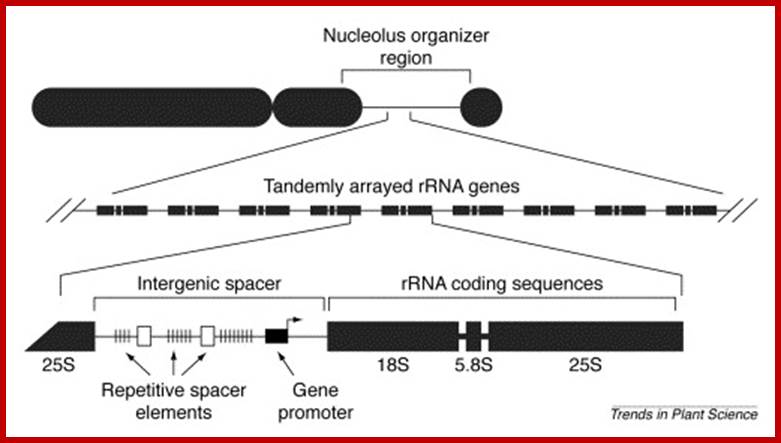
The mitotic metaphase chromosomes, under microscope appear as double stranded colored threads (after staining). Each of the longitudinal threads of a chromosome are called sister chromatids. They may be relationally coiled to each other or they may remain parallel to each other. But each of the sister chromatids are highly coiled and condensed structures. Such chromosomes show one or two constriction or non stainable gaps like regions. Based on the structure and functions, they are differentiated into primary constrictions and secondary constrictions. The former is called centromere and the latter is called nucleolar organizer.
Centromere (CEN):
It is recognized by its role in chromosomal movements. This region of the chromosome has a tiny granular structure acting as midpoint between the arms of the sister chromatids. At this region the metaphasic chromosome is still single stranded, that is why the two sister strands are still held to each other as a single chromosome. The position of the centromere varies but for the given chromosome it is always constant and characteristic. Under electron microscope, this region shows another structure called Kinetochore on either side of the CEN region.

http://www.nature.com/
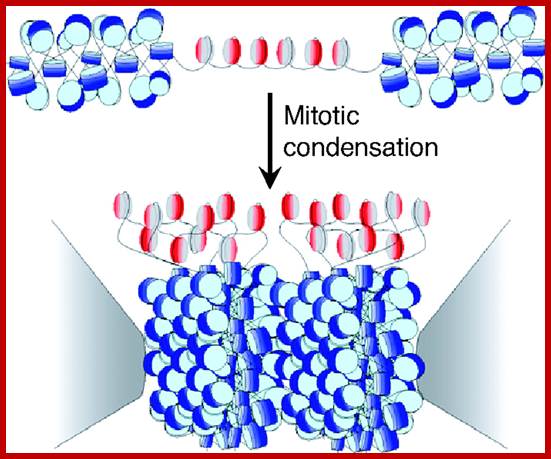
Chromosomes contain coiled coils of chromonema; The condensin complex (yellow circles) gathers the chromatin into lateral loops (blue), which are centered along a chromatid axis. Chromatin loop size determines the overall width of mitotic chromatids. Large loops are regularly spaced within and between genes to compact most of the genome into sausage-shaped chromatids. At specialized loci such as the centromeres, telomeres and rDNAs, chromatin loops are smaller, producing constricted regions. In the CEN region one can see the kinetochore structure with tractile fibers attached.
Kinetochore:
· Kinetochore is macromolecular structure
· Kinetochore consists of two proteinaceous commissural cups on either side of the CEN-centromere.
· Each cup like structure is made up of three layers of proteins.
· They are made up of specific proteins organized into such shape.
· On the inner surface of the cup, it is filled with a kind of matrix, which is attached to a specific sequence of DNA called centromeric DNA (CEN-DNA).
· The CEN-DNA has specific sequences, and such a sequence is repeated from few to several hundred times. This DNA belongs to a class of DNA called Highly Repetitive Class (HRC) of DNA.

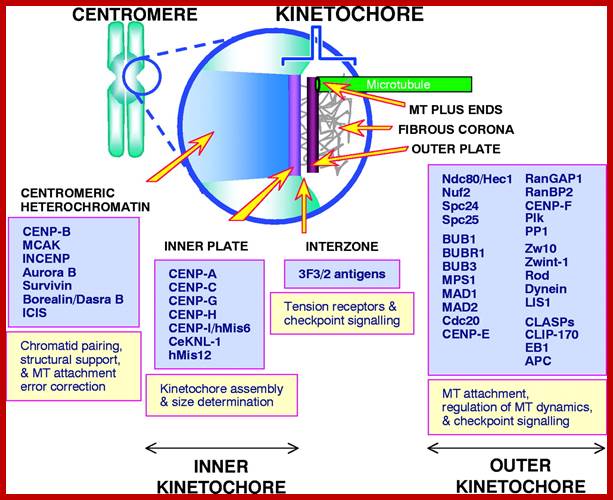
Organization of the animal kinetochore showing the locations of some of its protein constituents [updated from Pluta et al. (Pluta et al., 1995); Rieder and Salmon (Rieder and Salmon, 1998)].;http://jcs.biologists.org/
· On the outer surface of the cup like structure, fine hair like structures are present called coronary hairs. This region is also associated with specific proteins which are involved in chromosomal movement during cell division.
· Centromeric DNA that is associated with histones of special kind called CENP-A, CENP-B and CENP-C. The protein organization at this region is more complex, the number of proteins found varies.
· Added to this, there are ten to twenty bundles of microtubules appear as if they are radiating from the surface of the cup. In fact, during the organization of mitotic apparatus, these fibers extend into tractile fibers, which are responsible for the chromosomal movements.
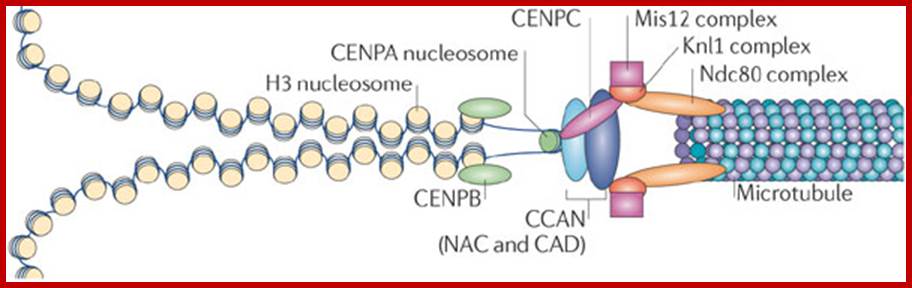
Kinetochore-The kinetochore is a large, multiprotein complex that is needed to link the sister chromatids to the mitotic spindle during chromosome segregation. The physical organization of the kinetochore into a trilaminar structure170, 171 is visible in electron microscopy images of vertebrate kinetochores, and the molecular composition follows this three-layer organization3, 5, 14, 147, 148, 172. The kinetochore forms on the microtubule plus end as a 'basket' of elongated molecules (namely Ndc80) that recruit the outer- kinetochore components of the KMN network (which comprises the Knl1 complex, the Mis12 complex and the Ndc80 complex) (see the figure). These outer-kinetochore proteins dangle from the expanded basket surface generated by Ndc80 and can interact with the chromatin and proteins of the constitutively centromere-associated network (CCAN). The purpose of the basket is to allow the outer-kinetochore components to move over a greater distance and increase the likelihood of encountering an unattached centromere. The geometry of the centromeric protein A (CENPA)-containing chromatin predisposes CENPA to be at the surface (see main text) and to recruit the CENPA-containing nucleosome-associated complex (NAC). The two halves of the kinetochore can then interact and form a stable attachment, connecting the chromosome to the microtubule. The kinetochore serves several important roles during chromosome segregation: it links chromosome movement to microtubule dynamics, monitors chromosome bi-orientation and serves as a site of catalysis for synchronizing chromosome segregation with cell cycle events. Jolien S. Verdaasdonk & Kerry Bloom; www.Nature .com
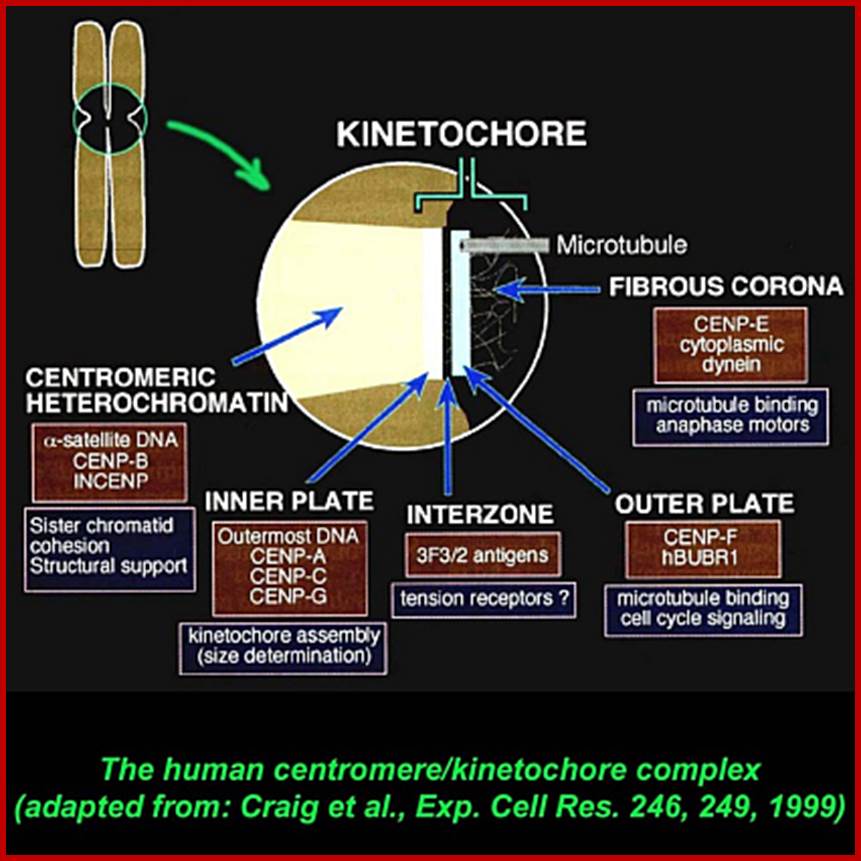
Craig et al, Exp.Cell res.246,249,199

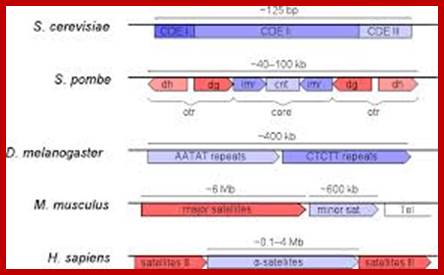
Organization of centromeric (blue) and pericentromeric (red) DNA in some eukaryotes. CDE, Centromere DNA Element; cnt, central core; imr, innermost repeats; otr, outer repeats; Tel, telomeric DNA.www.incenp.org
· In mitotic metaphasic chromosome two such kinetochore structures are present one at each side of the centromere, but in meiotic chromosomes only one at surface of the centromere. This is because only one kinetochore structure is present that to at the outer surface of each of the paired homologous chromosomes.
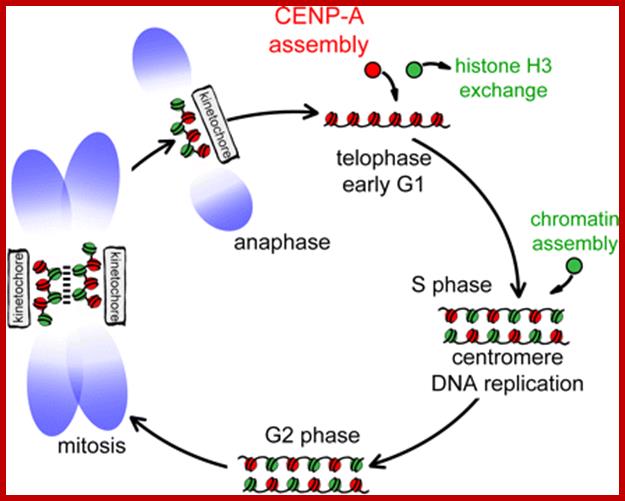
Centromeres direct chromosomal inheritance by nucleating assembly of the kinetochore, a large multiprotein complex required for microtubule attachment during mitosis. Centromere identity in humans is epigenetically determined, with no DNA sequence either necessary or sufficient. A prime candidate for the epigenetic mark is assembly into centromeric chromatin of centromere protein A (CENP-A), a histone H3 variant found only at functional centromeres. A new covalent fluorescent pulse chase
labeling approach using SNAP tagging has now been developed and is used to demonstrate that CENP-A bound to a mature centromere is quantitatively and equally partitioned to sister centromeres generated during S phase, thereby remaining stably associated through multiple cell divisions. Loading of nascent CENP-A on the mega base domains of replicated centromere DNA is shown to require passage through mitosis but not microtubule attachment. Very surprisingly, assembly and stabilization of new CENP-A– containing nucleosomes is restricted exclusively to the subsequent G1 phase, demonstrating direct coupling between progression through mitosis and assembly/ maturation of the next generation of centromeres. Lars E.T. Jansen,1, Ben E. Black,1,2 Daniel R. Foltz,1, and Don W. Cleveland1;www.mcb.berkeley.edu
Model for the kinetochore. CenH3 hemisomes (red/gray disks) are separated by extended linker DNAs and so are decondensed relative to surrounding heterochromatin (blue disks). Asymmetric CenH3 nucleosomes assemble in random orientations [CenH3/H4 (red) and H2A/H2B (gray)]. Only one unit of a CenH3-rich block is shown. During mitotic condensation, heterochromatin packs tightly as a result of its homogeneity. Intervening blocks of CenH3 chromatin cannot pack into this crystal-like structure because of its smaller size, long linkers, and heterogeneity in its relative orientation, resulting in extruded loops of uncondensed CenH3 nucleosomes that serve as the foundation for kinetochore formation. The flanking gray cones represent pericentric regions flanking the primary constriction. http://www.pnas.org/
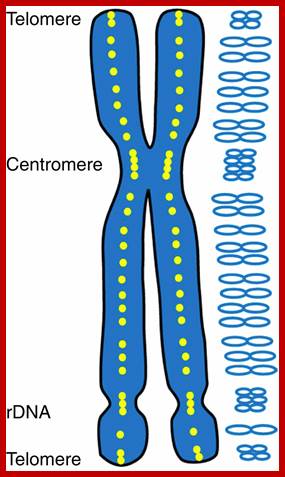
Telomeres:
Most of the chromosomes contain, yet another tiny granular structures. They are called as Telomeres. They are structurally very tiny and microscopic in size. They are found at extreme tip of chromosomes. Only with high resolution microscope it is possible to identify them. They appear to be fine sub-microscopic structures, stainable and found at each end of chromosomes.
https://exploreable.wordpress.com
· They provide stability to chromosomes.
· They prevent wrong end joining of broken chromosomes, where telomeric ends won’t rejoin to the broken ends.
· Telomeres have been isolated and their structural DNA has been characterized.
· The size of Telomeres is implicated in aging process or vice versa i.e. whether size of the Telomeres depends upon the age or Telomere size determines the age of the cell. Telomere size contracts and increases.
· Older and aged cells contain shrunken telomeric DNA. Active cells and cancer cells contain larger telomeric DNA.
· Telomeric DNA consists of a set of sequences which are repeated several hundred or more times, whose size can range from 5000 to 10, 000 bp.
· The repeat sequence is species specific, for example, Homo sapiens have 5’GGGATT’.
· As the Telomeric DNA has free ends, they are bound to be susceptible for exonuclease digestion, but the digestion is prevented by the Telomeric DNA structural organization in such a way, they are protected by the association a set of proteins.
· As the Telomeric DNA has sequences rich in GGGATT, the Gs can base pair with each other and generate a quartet structure, though not quadruplex form. Much such quartet structure can generate at Telomeric regions. Replication Telomeric DNA is explained elsewhere (DNA replication).
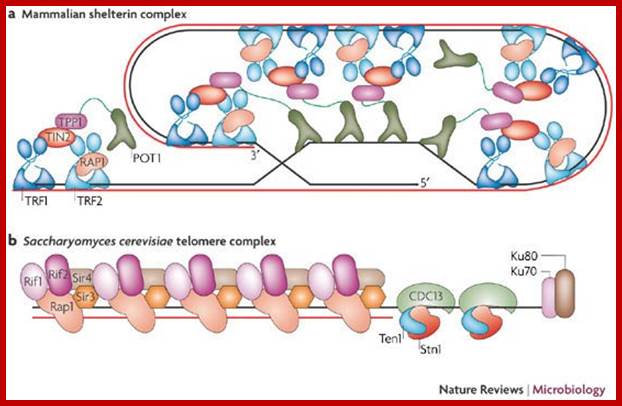
Telomeric protein complexes: a | Mammalian shelter in complex. TTAGGG repeat-binding factor (TRF) 1 and 2 are the predominant double-stranded TTAGGG-binding factors. Both proteins are stabilized by TRF-interacting nuclear protein 2 (TIN2), which interacts with TPP1 that, in turn, interacts with protection of telomeres 1 (POT 1), the single-stranded telomere-DNA-binding factor. TRF2 interacts with repressor activator protein 1 (RAP1) tightly and stoichiometrically. Functionally, sheltering negatively regulate telomere length. TRF2, POT1, TIN2 and RAP1 are also essential for telomere-end protection. b | Saccharomyces cerevisiae telomere protein complex. Rap1 is the predominant telomere duplex-DNA-binding factor, which tethers Rap1 interaction factors (Rif) 1 and 2 to negatively regulate telomere length, and silencing information regulator (Sir) 2, 3 and 4 to establish the telomeric heterochromatic structure. All these factors are essential for telomeric position effects (TPE), and Rap1 has a chromosome end-protection function. Cell division control 13 (CDC13) binds the single-stranded telomeric DNA and interacts with Stn1 and Ten1 to regulate telomere length. It also protects chromosome ends and has a role in TPE.
;Oliver Dreesen et al; http://www.nature.com/
www.blood journal.org
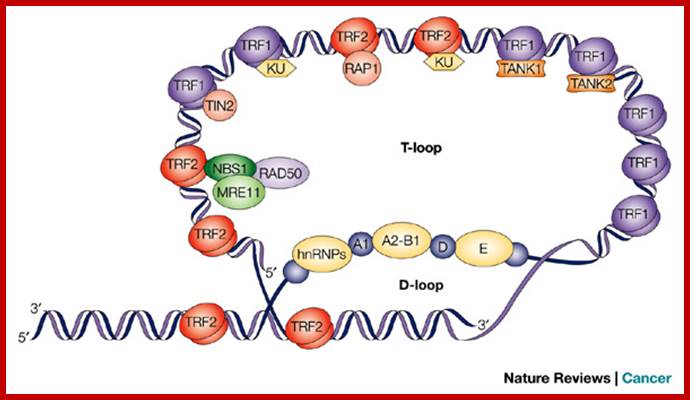
Schematic representation of putative telomere T-loop structure with telomere-specific binding proteins. The single-stranded DNA at the end of the telomere is able to invade and anneal with part of the duplex DNA (thereby forming a displacement (D)-loop) in the same telomere, with the overall result being a telomere (T)-loop7. Several proteins bind specifically to telomeric DNA, and these recruit other proteins to the chromosome end. Alex A. Neumann et al;www.nature.com
· The 5’ end of the Telomeric DNA, as seen under TEM is looped and its terminal end segment is inserted into the duplex and base paired with the lower strand, thus the both 5’ and 3’ ends are inside and are not exposed for exonucleases digestion.
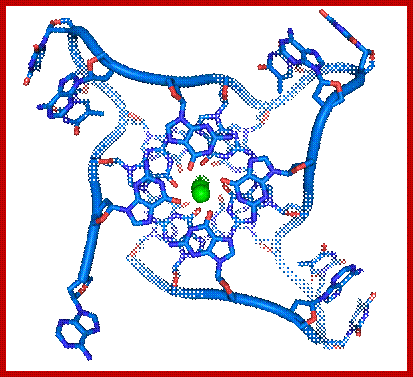
G in between repeat telomeric sequences pair with one another in the form of Quadruplex; www.studyblue.com
· Along with Telomere looks like a blob because of proteins, thus the ends are protected.
· Telomeric DNA often expands and shrinks in size during DNA replication for the ends don’t have any mechanism to fill the gaps after the removal of primers.
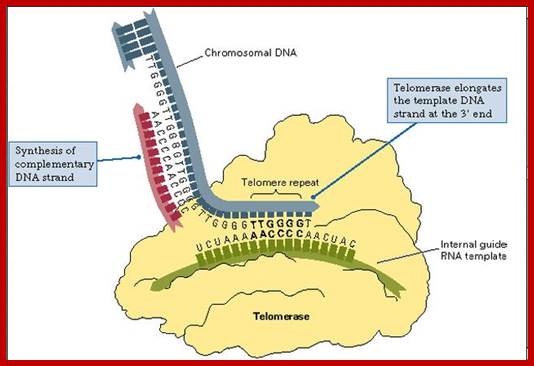
· But the DNA ends are replicated by specific enzyme complex called Telomerase.
· Telomerase consists of 120-130 ntds long RNA segment, an RNA dependent DNA polymerase (called Reverse Transcriptase), and few other TEF proteins.
· During early part of meiotic cell division, in animal cells, chromosomal ends with Telomeres are attached to nuclear membrane in a form called bouquet shape. What is the advantage having this configuration is not clear.

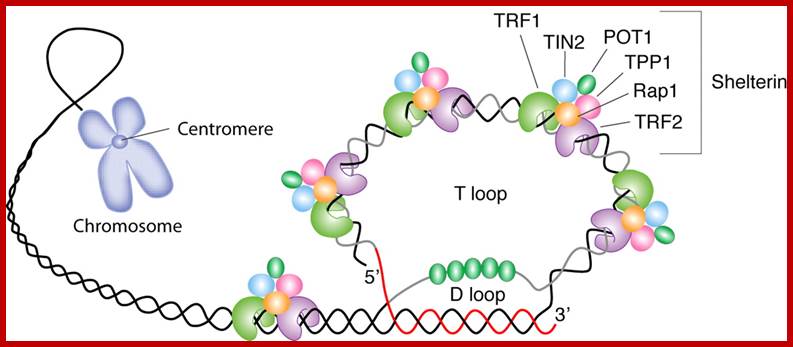
The fluorescence image shows the location of a telomere within a chromosome. Mammalian telomeres consist of TTAGGG repeats with a single-stranded 3' overhang of the G-rich strand. Specific protein complexes bind to the double- and single-stranded telomeric DNA. The components of the shelterin complex are shown in bold text. The single-stranded overhang can invade the double-stranded portion of the telomere, forming protective loops — such as t-loops with displacement loops (D-loops) — at the invasion site. The telomerase complex (which contains the telomerase RNA template and the reverse transcriptase TERT) interacts with the overhang and is regulated by shelterin and other telomeric proteins7. Other factors that can interact with telomeres are listed. Bidirectional arrows indicate interactions. Ramiro E. Verdun & Jan Karlseder www.nature.com
Nucleolar Organizers: These secondary constrictions like structures are found in one, two or more pairs of homologous chromosomes in a diploid set. Normally they are found just behind the Telomeric segments, but they can also be present any where in the chromosomal arms. But the position in a chromosome is species specific. This region is always associated with, nucleolus. It is at this region, the chromatin opens up and loops out into nacked DNA strands and provide template for transcriptional facility for the synthesis of ribosomal RNAs and for the organization of the nucleolus; it is this structure in which ribosome subunits assemble with rRNA and ribo-proteins into functional ribosomes. The size of the nucleolus can increase when the cell is very active and decrease when cell is in restive condition. During cell division as the nuclear membrane dissociates and the nucleolus also disappears, but as the Nucleus reassociates the nucleoli develop with transcription in rRNA genes.

http://palaeos.com/
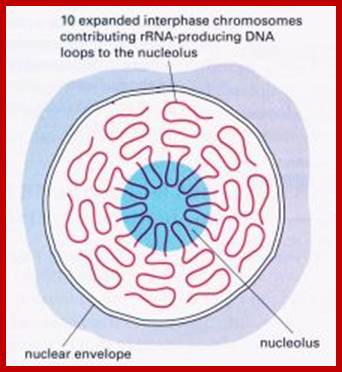
Central structure is Nucleolus hub if Pol I which transcribes nucleolar genes i.e rRNA genes, the transcripts are processed and required protein assemble into ribosomes; http://www.lifesci.dundee.ac.uk/
http://www.sciencedirect.com/
Chromomeres: Belling discovered rather observed, tiny granular structures in meiotic leptotene chromosomes. He called them as chromomeres. First he equated them to one chromomere to one gene. But now it is known, that each chromomere is a highly coiled and compacted region of chromonemal DNA. But under TEM or SEM such structures are not visible for the 30nm fibers are looped and bound to central scaffold. Each chromomere may contain one or more similar or different genes. The size of the chromomere is about 20000 to 60000 base pairs long or more. During gene expression, each of them open out into nacked DNA loops and they act as templates for the synthesis of rRNA. Lampbrush chromosomes are the par excellent example for demonstrating what are chromomeres and how they exist and when expressed how they look.
Satellites and Satellite DNA:
· The chromatin body found beyond secondary constriction is often called SAT or satellite.
· This region looks like a blob and it is totally lacking in thymidilic acid i.e., Sine Acido Thymidine, so it is called SAT. It also means the DNA in these regions is rich in GC content.
· Chromosomes containing these structures are called SAT-chromosomes; they act as markers in karyotype studies.
· Satellite can also be located interstially. The function of these structures is not clearly known. Whether or not the SAT structures are artifact, the DNA on high-speed centrifugation exhibits some minor bands on either side of main band. Such bands are called satellite DNA, each of which shows characteristic sequence identity.
· These SAT DNAs are made up of short sequences, which are repeated several hundred to thousand times; Based on the sequence length and the number of repeats they are classified into Mini-satellite DNAs and Microsatellite DNAs.
· They are made up of simple sequences, but repeats.
· These DNA segments can be used for DNA profiling or Genetic finger printing.
· DNA profiling uses PCR technology for amplifying the DNA using specific primer sequences.
· DNA profiling has helped in solving parental disputes, Phylogenic identification and criminal investigating.
Chemical Composition of Chromosomes:
Fortunately, recent techniques have come very handy for the isolation of pure chromosomes, by pulse field gel electrophoresis (PFGE), which can be subjected to chemical analysis or they can be mounted on an electron microscopic grid for studying the morphology.
Chromosomes are basically made up of proteins found in chromosome is two times more than the total mass of DNA. However, the total RNA associated with chromosomes is less than 10% of the total mass of DNA and most of the RNAs are in the form of nascent transcripts. With regard to proteins, chromosomes contain two classes of proteins called nonhistones and histones and in some cases protamines are also found.
Nonhistones: All chromosomal proteins, other than histones, are grouped under non histones (NH). The number of NH proteins found in chromatin varies from 750-2000 different kinds. Some of them are basic proteins and others are either acidic or neutral proteins. In fact, depending upon the electrophoretic mobility and chemical content, they have been classified into A, A2, B, C and D groups. The mol. wt of such proteins varies from 10 KD to many million Daltons. Nonetheless, the kind and number of non-histones present varies from tissue to tissue and species to species. Appearance and disappearance of some non histones during different developmental stages is very interesting. The most abundant non-histones are Topoisomerases and high mobility group of proteins (HMGs)
A number of non histones help in the structural organization and stability of chromatin fibers, but a large number of them have regulatory functions. DNA polymerases, RNA polymerases, ligases, etc are very important enzyme components of non histones. Many others help in folding and unfolding of chromonemal fibers during replication and transcription. Proteins like HMG (highly mobile group of proteins) and others act as receptor proteins for various stimuli. Steroid hormone receptors for estrogen and ecdysone belong to non histones, which regulate the expression of genes. Whole lot of them is involved in gene regulation.
Histones: Histones are mostly basic proteins for they contain greater amount of amino acids like lysine and arginine. They are found in equal ratios with the total mass of DNA.
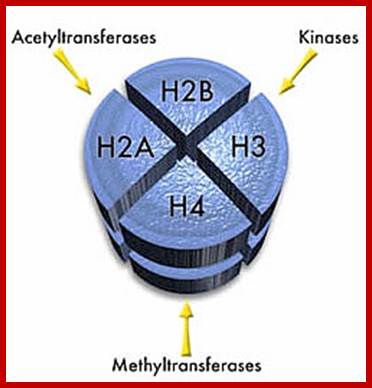
Based on the composition and sequence of amino acids and electrophoretic mobility, five classes of histones have been recognized. They are H1, H2A, H2B, H3 and H4. The H1 are highly rich in lysine and their composition varies from tissue to tissue and from species to species. But H2A and H2B are slightly rich in lysine but show little variations in their chemical composition. On the contrary, H3 and H4 are very rich in arginine and lysine; most of these proteins are highly conserved in most of the species from alga to whale. Histones have an important function in structural organization of chromosomal fibers.
Protamines: They are another class of proteins which are rich in arginine. But they are invariably found in spermatids of fishes, birds, squids and mammals. They play a very important role in the organization of spermatid chromosomes into very compact and tightly coiled structures
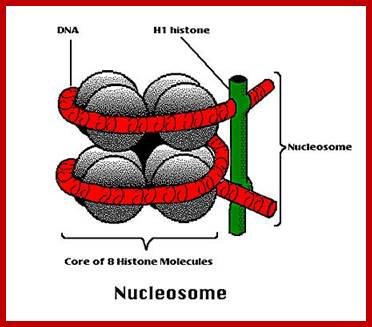
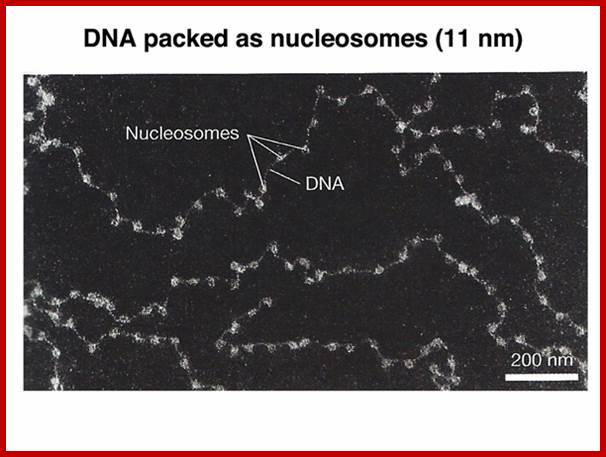
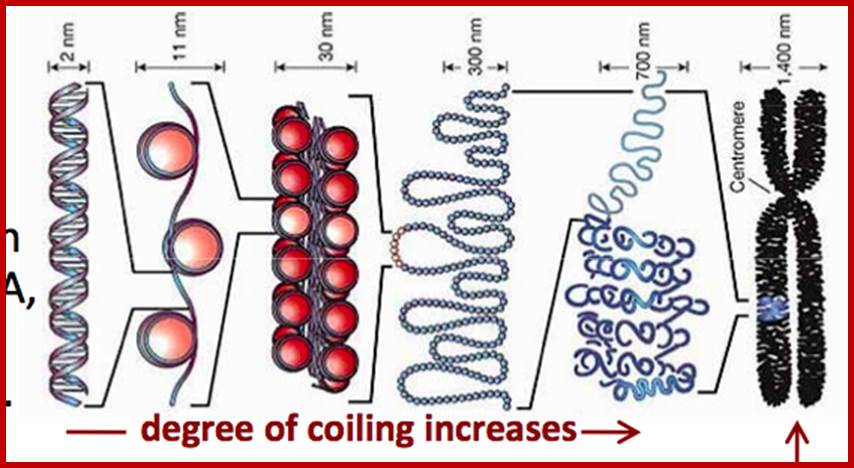
Nucleosomes: If chromosomes are isolated in a low salt solution; they disperse into a fine thread which looks like a string of beads of 11 nm thickness. The bead like structural units is referred to as Nucleosomes, which act as fundamental structural units of chromosomes. However, the inter bead length in each thread is constant. Moreover, each chromosome contains only one long molecule of DNA duplex organized into nucleosomal threads in association with histones, thus histones are the fundaments structural units that organize DNA in to stable structure.
https://www.studyblue.com

Image05.jpg
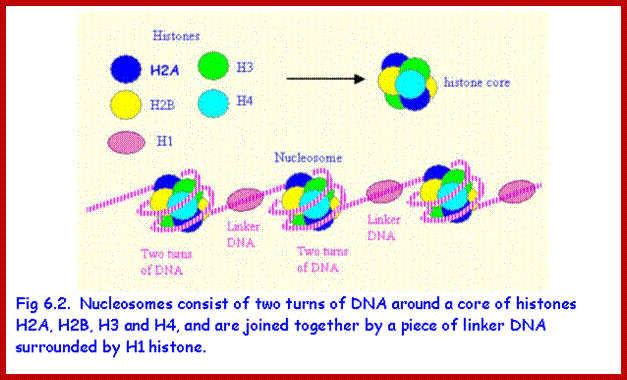
Nucleosome consists of bundled two pairs of Histone H2A,H2B, H3 and H4; wrapped around two turns of DNA; http://staff.um.edu.mt/

www.faculty.tru.ca
http://www.discoveryandinnovation.com/BIOL202
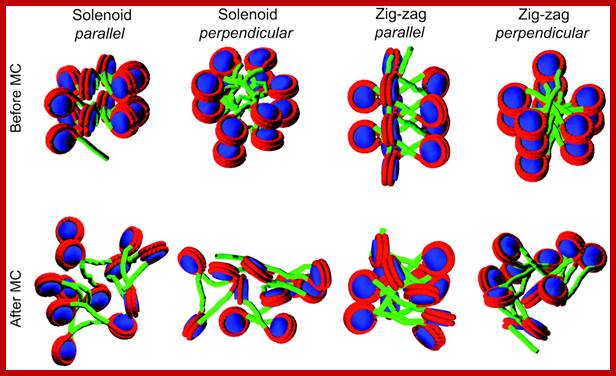
Solenoid Fibers:
Neutron scattering and electron dichroism techniques of nucleosomes reveal that each nucleosome is made up of an octamer complex of 2H3, 2H4, 2H2A and 2H2B histone proteins of which 2H3 and 2H3 act as the core proteins. To begin with, the interaction between histone H2A, H2B and H3 and H4 results in the formation of tetramer complexes called half nucleosomes, later they join with each other to form an octamer unit (Fig. 3A-30). Such octamer complex appears like an ellipsoid disc of 11 nm x 6 nm dimensions. The DNA duplex coils round such discs one three fourth. The total length of DNA required for two such coils around the octamer complex is about is 142-150 nucleotide base pairs, which indicates the DNA packing ratio is 6 (i.e. 67 nm / 11 nm).
The association of DNA with histone-octamer complex is greatly facilitated by a non histone acidic protein of mol. Wt of 29 KD called nucleoplasmin.
Individually, these proteins do bind to DNA directly, some bind to histones and other accessory proteins but they bind to histone complex and bring about conformational changes in the protein which help in the interaction between phosphate groups of DNA and amino groups found at the outer surface of histone proteins. The DNA association with histones is so tight; it resists even endonuclease digestion of DNA to a great extent. The length of the DNA found in between two nucleosomes is bout 80 – 114 nucleotides called linker DNA. Association of DNA with histone octamers has no sequence relationship, but there can be a bias, nevertheless the coiling of the DNA around histones and its ends to be in super coiled state.
H1 plays a role in this compaction; www.proprofs.com; www.quizlet.com
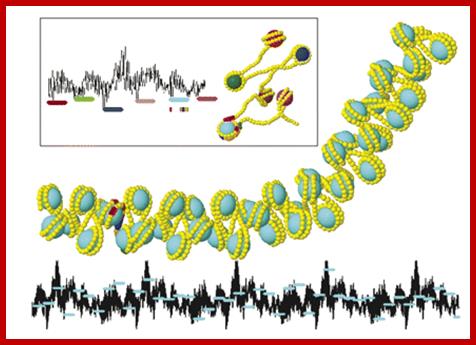
The Interactive Chromatin Modeling (ICM) web server allows users to investigate relationships between nucleosome positioning, DNA sequence properties, and the structure of chromatin. The six positioned nucleosomes of the mouse mammary tumor virus promoter, MMTV, yield a rather extended structure (insert). In this case the nucleosomes are well separated. Local minima in a sequence based intra-nucleosome stability landscape (black line) properly predict positioning. For condensed chromatin (main image) intra-nucleosome stability yields to inter-nucleosome interactions. Not all nucleosomes can occupy local minima on five repeats of the MMTV. Some linkers contain bent DNA resulting in bent chromatin. http://nar.oxfordjournals.org/

http://www.diffen.com/
www.studyblue.com
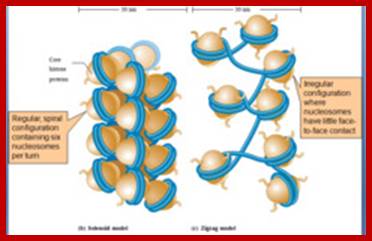
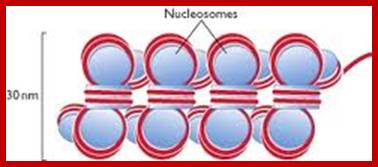
The DNA that is found in between two nucleosomes is called linker DNA, whose length varies from 80-110 base pairs. Histone protein called H1 and some non histone proteins bind to this linker DNA and bring about the compaction and coiling of nucleosomal threads into 30 nm thick fibers called solenoid fiber, which forms a basic chromonemal fiber. The solenoid fiber consists of coiled nucleosomal thread with 6 nucleosomes per coil. Or this nucleosomal thread in association with H1 histones can be compaction in zig-zag fashion, which also looks like solenoid fibers. The packing ratio in the solenoid fiber is 10.

Folded and Coiled Chromatids: The solenoid fiber of 30 nm thickness further undergoes folding and coiling to produce cytologically visible threads called Chromatids. Though the folding of 30 nm solenoid fiber appears to be random, it is actually determined by specific sites and specific folder proteins, which are believed to be non histone proteins. The site and the length at which the 30 nm fiber folds vary, nonetheless it cannot be said it is predetermined. The bottom ends of the loops are attached to a protein skeletal work called scaffold. The scaffold protein identity has been established. Such folding results in the long solenoid fibers into short and compact chromosomal strands, which themselves appear as spirally coiled structures. The folding is greatly aided by metallic ions like calcium and copper. But some non histones are very important in the folding process for they act as a scaffold.
Euchromatin and Heterochromatin:
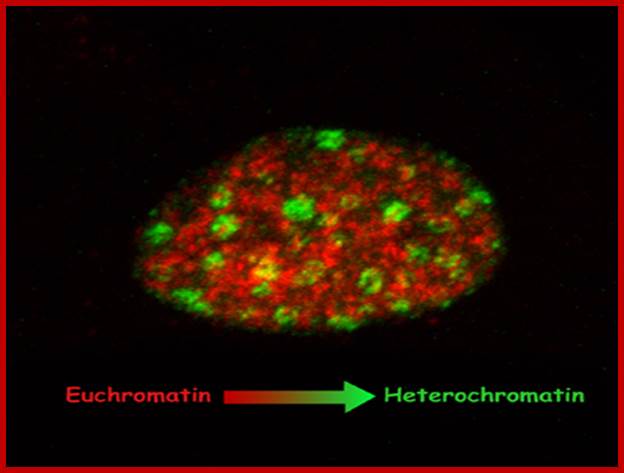
Differential staining shows Euchromatin and Heterochromatin region of chromatin.
The folding of solenoid basic fibers
is not uniform. In certain regions the folding is extremely compact and others
regions are not so. Highly compacted regions are called heterochromatic
regions and the others are called euchromatic regions; they can be resolved by
differential staining by using Giemsa. Heterochromatic regions exhibit two types’
namely constitutive heterochromatic regions and facultative heterochromatins.
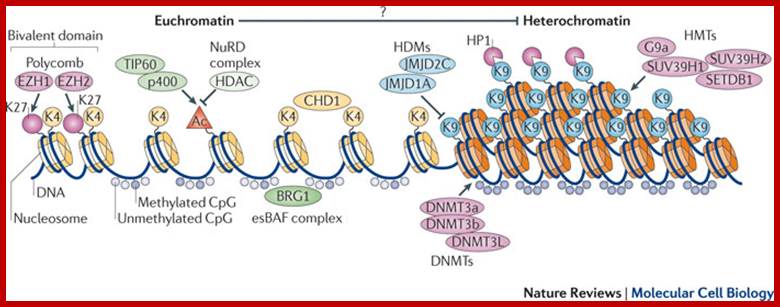
The balance between euchromatin and heterochromatin in ES cells-Several epigenetic regulators orchestrate the open chromatin state of embryonic stem (ES) cells and set the stage for the transcriptional network. Relevant epigenetic marks include histone modifications and incorporation of different core histone variants (yellow and orange cylinders) that alter access and efficiency of the transcriptional machinery. The main histone marks, the active H3 tri-methylated on Lys 4 (H3K4me3) and the repressive H3K9me3 and H3K27me3 (represented by the circles K4, K9 and K27), are positively regulated by specific histone methyltransferases (HMTs; including G9a (also known as EHMT2), SUV39H1, SUV39H2 and SETDB1) and negatively regulated by the respective histone demethylases (HDMs; including jumonji domain-containing 2C (JMJD2C; also known as KDM4C) and JMJD1A (also known as KDM3A)). Active (K4) and repressive (K27) marks can be present in the promoter regions of developmental genes to prevent their expression while allowing rapid activation by transcription factors such as the polycomb proteins Enhancer of zeste homologue 1 (EZH1) and EZH2 (termed bivalent domains). Histone acetylation also marks active chromatin, and the acetyl group (the orange triangle, Ac) can be added through complexes such as TAT-interacting protein of 60 kDa (TIP60; also known as KAT5)–p400 and removed by histone deacetylases (HDACs), which can be part of repressive complexes such as the nucleosome-remodelling (NuRD) complex. DNA (dark blue line) methylation is typically present on CpG islands in promoter regions and heterochromatin (marked by H3K9me3 and heterochromatin protein 1 (HP1)). DNA can be hypermethylated, as a result of the action of DNMTs, such as DNMT3a–DNMT3b or DNMT3L, but in euchromatic regions DNA is generally unmethylated. Chromatin-remodelling proteins such as chromodomain helicase DNA-binding 1 (CHD1) and BRG1 in the ES cell-specific BRG- or BRM-associated factor (esBAF) complex may regulate the open chromatin state, possibly by contributing to boundary determination between euchromatin and heterochromatin. There is growing evidence that the formation of euchromatin can repress the establishment of heterochromatin nearby (as it has not been confirmed in ES cells, this is denoted by a question mark). Alexandre Gaspar-Maia et al : www.nature.com
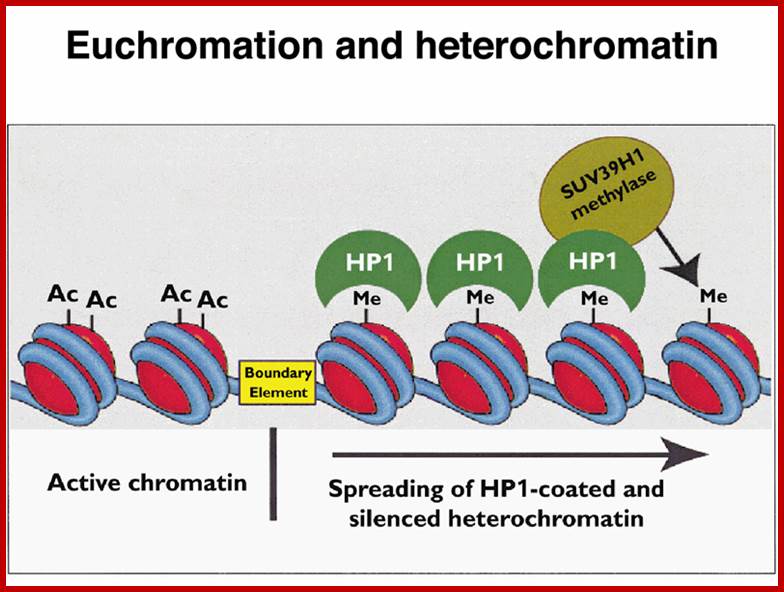
http://www.discoveryandinnovation.com/BIOL202
The constitutive heterochromatin regions are always restricted to certain loci. For example, on either side of centromere, the chromatin is heterochromatic, which never unfolds except at the time of the replication. The DNA present in such regions is distinct and settles as a satellite DNA. Telomeric region also shows constitutive heterochromatin. Such regions can be identified by specific stains called C and G Bands. On the contrary, the loci of facultative heterochromatic regions in a particular chromosome show variations; in the sense their loci shift from one position to another in different tissues and at different stages of development of an organism. Thus, facultative heterochromatic regions are mobile.
Transcriptional studies indicate that the heterochromatic regions are transcriptionally inactive, while the Euchromatin regions are active. Such transcriptionally active bonds can be identified as G bonds. The above observation explains the differential transcription, which varies from tissues to tissues and also varies from stage to stage of development. The active gene expression takes place is Euchromatin and the gene expression is mostly shut off in heterochromatic regions.
The folded chromosomes show some interesting features like folding sites and folded loops. If metaphase chromosomes are treated with dextran sulphate and heparin at low salt concentrations, most of the histones are dissociated and removed. This results in the release of DNA in the form of free loops. It almost looks like the spilled out DNA from chromatin. The length of loops varies from 20-60 nm consisting of 60-180 kilo base pairs which when compacted by histones and non histones gives 200-600 fold compactions. It is evident from the above observations that individual folds with certain loops represent one or more transcriptional units. Such conclusions stem from the fact that in Lampbrush chromosomes and salivary gland chromosomes at certain regions, DNA molecules loop out into actively transcribing regions. The comparison of the lengths of DNA loops that are free from histones due to disruption of DNA associated proteins with dextran sulphate treatment and the DNAs loops develop in lamp brush or Balbiani rings of salivary gland chromosomes vary in size, which strongly suggests that the specific foldings and loops are specific transcriptional units.
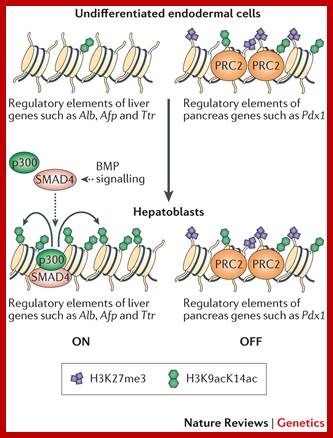
Epigenetic 'pre-patterning' for lineage specification. In multipotent endodermal cells, regulatory elements of liver-specific genes and pancreas-specific genes are pre-patterned with distinct chromatin marks. Both the active histone H3 lysine 9 and lysine 14 acetylation (H3K9acK14ac) marks and the repressive H3K27 trimethylation (H3K27me3) marks are enriched in the regulatory elements of pancreas-specific genes (such as pancreatic and duodenal homeobox 1 (Pdx1)), but they are either low in abundance or undetectable in the regulatory elements of liver-specific genes (such as albumin (Alb), alpha fetoprotein (Afp) and transthyretin (Ttr)). In response to bone morphogenetic protein (BMP) signalling, SMAD4 recruits histone acetyltransferase p300 to the regulatory elements of liver-specific genes to stimulate histone acetylation and to induce hepatic specification. Polycomb repressive complex 2 (PRC2) maintains the level of H3K27me3 in the regulatory elements of pancreas-specific genes to prevent pancreas specification. Taiping Chen ,& Sharon Y. R. Dent; www.nature.com
http://www.rsc.org/

Scientist built synthestic chromosomes;
www.article.wn.com
Synthetic Chromosomes: In Chicago - An international team of scientists has built a yeast chromosome from scratch; the latest step in the quest to make the world's first synthetic yeast genome, an advance that would lead to new strains of the organisms to help produce industrial chemicals, medicines and biofuels.
Instead of just copying nature, the team did extensive tinkering with their chromosome, deleting unwanted genes here and there. It then successfully incorporated the designer chromosome into living yeast cells, endowing them with new capabilities not found in naturally occurring yeast.
“It is the most extensively altered chromosome ever built,” said Jef Boeke of New York University's Langone Medical Centre, who led the effort. The findings were published in an online edition of the journal Science.
While other teams have synthesized bacterium and viral DNA, Boeke's is the first report of a synthetic chromosome in a eukaryote, an organism whose cells contain a nucleus, like human cells.
The achievement, which took seven years, involved the use of computer-aided design to construct one of 16 chromosomes in brewer's yeast, known scientifically as Saccharomyces cerevisiae.
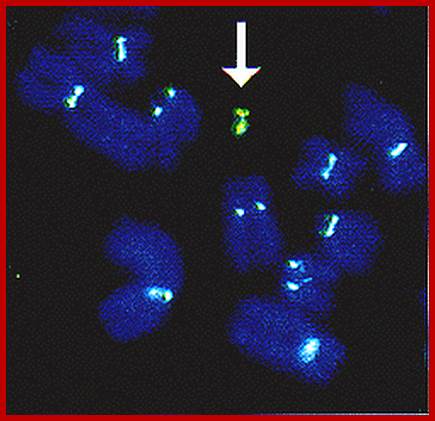
The overall structural organization of chromatin shows that histones basically initiate the folding of long DNA molecules into compact bead shaped threads called nucleosomal threads. Later such threads undergo compact coiling into solenoid fibers and then into folded and coiled chromonemal fibers. The bizarre folding pattern, observed under electron microscope is not random as seems to be but there is a method in randomness. Scientists’ have created artificial chromosomes by using Yeast artificial vector (YAC). The arrow shows the presence of such synthetic chromosomes. Such designed chromosomes will come handy for therapy.
SPECIAL TYPE OF CHROMOSOMES
i) Salivary gland chromosomes:
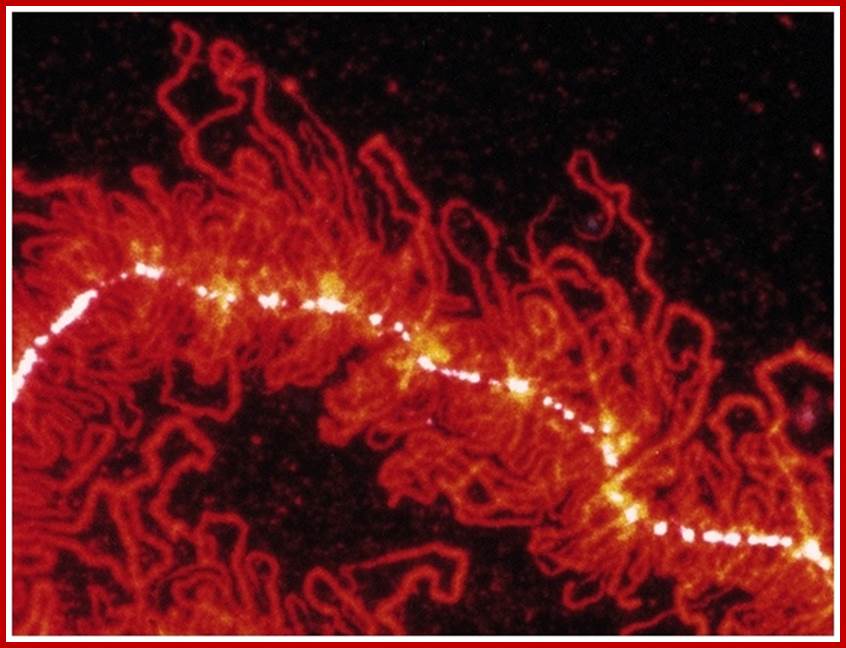
In the salivary glands of larvae belonging to certain insects like Drosophila, mosquitoes and other dipteran insects, little earlier to transformation of larvae into pupae, the normal chromosomes transform into giant chromosomes. During such transformations somatic chromosomes undergo synaptic pairing between homologous. Each of these chromosomes repeatedly duplicate without separation and end up in the formation of 1080 and odd strands (endopolyploidy). Such chromosomes are also called polytene chromosomes. Because of these changes, salivary gland chromosomes appear as long and thick structures.
Heterochromatin and Euchromatin Bands;
Euchromatin and Heterochromatin bands: The salivary gland chromosomes contain many darkly stainable transverse bands, alternating with lighter bands. The former is heterochromatin and the latter is euchromatin. The heterochromatin bands are the regions where the chromatin is tightly packed and the genes are believed to be inactive in such regions. On the other hand, the euchromatin bands are coiled normally but loosened and the genes are active in these regions.
Chromosomal Puffs and Balbiani Rings: During larval transformation, certain loci in the giant chromosomes become active. At this stage, the looped chromatins unwind and open out into fine loops of various sizes. At each loci, all the thousand and odd chromatin strands open out into long loops; thus it looks like a puffed region.
The open loops are nacked DNA molecules engaged in active transcription. The synthesis of the required RNAs once translated, the products are used for the transformation of larvae into pupae. Such giant loops with active transcription and translation are called Balbiani rings, named after the discoverer. The puffs and Balbiani rings are the cytological expression of differential gene activity. The site (loci), size and the number of puffs vary from stage to stage during the development of larvae.
ii) LAMPBRUSH CHROMOSOMES:
These are another class of special chromosomes. They are found in the Oocytes of frogs, salamanders, certain birds and fishes. While oocytes are undergoing meiosis, homologous chromosomes start pairing and later express in chiasmatic configuration. It is during these stages the homologous chromosomes, still held by chiasma, elongate considerably. At the same time a large number of chromomeres (at least 500) per chromosome uncoil and open out into fine loops from each of the double stranded chromosomes.
www.forum.daffodilvarsity.edu.bd
Chromomere loops: Each of these loops is nacked DNAs and they are transcriptionally active. Such gene activated products arc required for the development of eggs into future embryo. Lampbrush chromosome is yet another example for the cytological manifestation of differential gene expression. As most of these chromosomes look like bottle brushes (once used to clean lamp glasses), they arc called as Lampbrush chromosomes.
SEX DETERMINATION
A majority of plants and animals exhibit sexual mode of reproduction. Some of them are bisexual (hermaphrodites) and some of them are unisexual. In higher animals unisex is a predominant feature, where male and female sexes arc distinguished by characteristic structural and functional features. Even many plants exhibit this kind of sexual dimorphism.
Sex chromosomes and Autosomes:
Cytogenetic studies, in the past fifty years or so, have established that the sex of on organism, in its early development is determined by special chromosomes called Sex chromosomes or Allosomes, such as X and Y. All other chromosomes are called Autosomes (A). All unisexual animals including Insects, Fishes, Frogs and mammals including human beings show XX and XY mechanism of sex determination. Even some unisexual plants like Melandrium, Coccinia, Sphearocarpus etc. show XX and XY mechanism. However, certain variations are also found as in the case of birds.
(1) Sex determination in human beings:
The diploid number of Homo sapiens is 46. In females, 2n = 44 A + XX is the chromosomal pattern, but in males 44A + XY is the chromosomal composition.
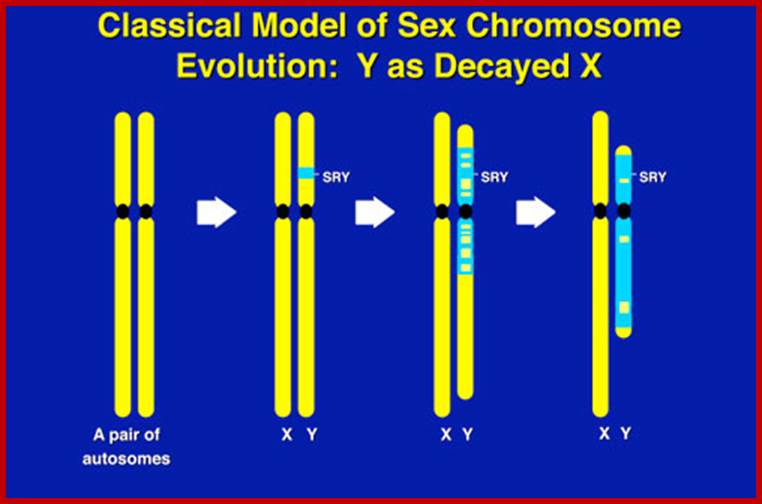
The X chromosome in males is euchromatic (genetically active) and Y chromosome is heterochromatic; assumed to be genetically inactive except for few genes which are active, ex. Sry. In fact, it is now established that the male determining gene is located in Y chromosomes and the female determining genes on X chromosomes.
There are cases where the chromosomal composition in certain patients was found to be 44A + XO i.e. one of the sex chromosome is missing and the sex is female. In another case 44A + XX shows male sex. This is because; in the first case the Y chromosome is completely missing. So, the X chromosome determines the sex of an individual as female. But in the second case (44A + XX), a segment of Y chromosome containing male determining gene(s) is somehow translocated to one of the X chromosomes. In this special case, inspite of the absence of Y chromosome the sex of the patient is male, because the translocated segment of Y chromosome contained sex determining gene. This clearly establishes it is XX and XY combination that ultimately determines the female & male sexes respectively. The same is true with many higher animals and also some plants.
Out of two X chromosomes in females one is euchromatic and the other is totally heterochromatic with the exception of 6-7 genes that escape X-chromosome inactivation and it appears as a barr body in females. In males the barr body is absent. If by chance a barr body is identified in males, then this chromosomal composition is 44A + XXY, and it is an abnormality.
But in birds 2A + ZZ determines male sex and 2A + ZW determines female sex.
2. Sex determination in Drosophila: In Drosophila, mosquitoes and other dipteran insects, the sex determination is by XX and XY mechanism, but with a difference when compared to the sex determination of human beings. Its 2n number is 8. If 2n = 6A +XX; it is female. If 2n = 6A+XY then it is male. But if an insect has 2n = 6A with only one X chromosome, then the insect develops as a male. This is due to the balance between autosomal number and sex chromosomes; here male sex is determined inspite of the absence of Y chromosomes.
6A+XX = Female 6A 4- XXY = Super female
6A+XY = male 6A + XYY = Super male
6A L XO = Male
Molecular mechanism of sex determination in Drosophila has been more or less elucidated. The genes involved in the processes are sXl, dsXl and few other related genes. The alternate splicing of the said gene transcripts play a significant role in determining the sex of the organism at an early stage of tissue differentiation.
3. Sex determination in Honey bees: In honey bees, the queen bee lays eggs and the worker bees collect honey. The queen bee is always diploid and it alone is capable of laying eggs. If the eggs fertilize with a sperm, the animal develops into females. On other hand if the haploid egg remains unfertilized, it develops into a male donor bees or slave bees.
4. Sex determination in bacteria: In bacteria, males are slightly bigger in their cell size than their female counterparts. The male sex of bacteria is determined by the presence of an episome containing fertility factor. If the said factor is absent, then the bacterium acts as female.
5. X-chromosome Inactivation: After fertilization the zygotic cell undergoes cell divisions and cell differentiation. During early stages of cell divisions in females both XX chromosomes are active, but with more cell divisions, one of the two X-chromosomes randomly under go inactivation, thus some cells contain X from pa’ inactive and in some cell x-chromosomes from ma’ remains inactive, thus the gene expression produces a mosaic of characters.
X-chromosome inactivation is due to the production of Xist RNA which covers the entire chromosomal thread and localizes in bands. The Xist RNA is expressed from Xic gene loci. The RNA has a cap and a poly-A tail but no coding information and it remains untranslated, for it does not move out of the nucleus.
SEX LINKED INHERITANCE
Similar to autosomes, sex chromosomes particularly X & Y chromosomes also carry expressible genes.
As sex chromosomes determine the sex of an organism, the genes found on sex chromosomes, as a linkage unit, also express their phenotype which is restricted to a particular sex of an organism. Such type of inheritance and expression limited to only one or the other combinations of sex chromosomes is called sex linked inheritance. So certain characters are inherited to either female or male sexes.
X-linked characters: Certain characters like color blindness, hemophilia muscular dystrophy in human beings, red and white eye color in Drosophila melanogaster; and black and barred plumage in poultry are some of the examples of sex linked, specifically X linked characters.
Y-linked characters: The Y chromosome also contains many genes in human beings but they don't express. Even if expressed, it is difficult to identify. But recent investigations do indicate that Y chromosomes also carry genes which express only under certain intracellular environment A gene for hypertrichosis i.e., hairs on external pinna, male sex determining factor SRY, HLA - antigen and overall height controlling factors have been assigned to Y chromosomes.
PATTERN OF SEX-LINKED INHERITANCE:
Eg. Color blindness:
Persons having color blind trait cannot distinguish red color from the green color. This character is controlled by a receive gene called “c”. The normal wild type gene is "C" gene. These genes are located on X chromosomes. If the female is heterozygous for this character i.e., “Cc", her color sensitivity is normal. On the other hand, if she is homozygous for color blind genes "cc" she is color blind. On the other hand, if a male carries a "c” gene on X chromosome; he is color blind; it is because the Y chromosome docs not carry any normal allele for c’ and it is genetically inactive.
Criss-cross inheritance: If the female, heterozygous (Cc) for color blind gene, marries a normal male, (Cy), the probability of getting female to male children is 50:50. Among males, 50% will be color blind and other 50% normal. But all the females will be normal Follow the diagrammatic representation below.
Instead, if a heterozygous (carrier) female (Cc) marries a color-blind male Cy: 50% of the females are color blind and 50% of the male children will be color blind and other male and female children are normal.
The same pattern of sex-linked inheritance is also found with respect to hemophilia gene.
Sex influenced characters:
Not all phenotypic characters expressed in females or males show sex-linked inheritance. For example, in human species beard, moustache, hair development on the chest, voice cracking, masculinity etc., are restricted to males. Similarly, non development of beard, moustache, chest hairs, non cracking of voice and development of breast etc are restricted to females. The above said characters, restricted to a particular sex, are expressed not at the early childhood but manifested at the adolescent stage. Such characters are called sex influenced or secondary sexual characters.
The genes responsible for the expression of such characters are not found on sex chromosomes. However, the expression of such characters is under the influence of sex hormones and the age. Even masculinity and feminism are secondary sexual features.
Another good example is milk production in cattles. The genes responsible for milk production are present in both males and females, but they are expressed only in females because the female hormones influence the genes to produce milk.
Sex limited characters: Development of horns mostly in male sheep, formation of characteristic comb and feathers in male cocks and baldness mostly in male humans ere actually sex limited characters.
Chromosomal Remodeling and Gene expression:
Gene expression:
Genes are units of heredity; chemically and physically consist of a length of DNA encoded with regulatory elements and sequences for coding a polypeptide chain or RNAs. Eukaryotes have at least three basic types of genes for they are transcribed by different set of RNA Pols. RNA Pol I transcribes exclusively rRNA genes; RNA pol II transcribe all mRNAs and few noncoding RNAs too; RNA Pol III transcribes genes involved in producing tRNAs, 5srRNA gene and few Sc RNAs like 7sl RNA genes.
Most of the RNA pol I and RNA pol III transcribed genes are common to all tissue types, but Genes transcribed by RNAPol II differ from one tissue type to the other, however 70% of the gene transcribed are same, for they act as house keeping genes. In human body there are at least 320 different cell types, each have their own characteristic shapes, structure and the gene expression is specific to the cell type along with house keeping genes. In additions several genes express in response to internal or external signals. Differential gene expression can be detected by using mouse system. First total mRNA is isolated and converted to ds cDNA by reverse transcriptase. These are placed on micro plate, least 100 spots (not enough but to test); and denatured to provide ssDNA. Isolate mRNA form brain and kidney, differentially label them (to detect brain and kidney transcript) while they are converted to cDNA. Such cDNA are used to hybridize the spotted cDNA from the liver. Majority of the cDNA from livers are hybridized by brain and kidney mRNAs but, some are not hybridized to liver transcript. It means the cDNA of liver not hybridized to kidney and brain are different, but those hybridized are common for all the tissues. This can be more exemplified by using DNA microchips and performing microarray hybridization.
Assembly of BTA (PIC):
Transcriptional complex that initiates protein coding genes is a multiple subunit complex that forms BTA or PIC. It binds to TATAA, InR and DPE elements of the promoter. But the eukaryotic promoter elements for different genes have different regulatory elements, such as genes containing TATAA, InR and DPE elements with few common upstream elements, but there are promoters which are TATAAless but InR plus and DPE plus, in addition there are a large chunk of genes contain just DPE but no TATAA and no InR regions. However near by upstream elements are found by factors such as CAAT and GC rich and GATA that bind upstream act as activators, which form a transcription initiation complex. Factors like activators and enhancers proteins, which are located nearby within -100 to -200 and Enhancers at -400 to -1000 upstream or + 400 to +1000 down stream elements bind to such regulatory regions in gene specific manner (house keeping gene expression gene constitution is different) and activate specific genes.
Assembly of Pre-initiation complex (PIC) or Basal transcriptional apparatus (BTA) or GTFs complex-general transcriptional factors, is not automatic, they are recruited by activator-co activators complexes that bind at upstream elements. Enhancer bound factors; in most of the cases enhance the transcriptional efficiency by 100 to 200 times. They are the same structural and functional features similar to that UAS element of Yeast regulatory elements.
Assembly at TATAA starts of course, triggered or assisted by other upstream factors, with the binding of IIF D complex which consists of TBP and TAFs. TBP is a monomer protein with horse shoe shape or horse stirrup shape binds to TATAA at the minor groove and induces some structural changes in the DNA- induces bending and slight expansion of the DNA diameter from 19- 20 A to 25A. Bending and opening of the DNA helical structure is very important for forming transcriptional initiation and Bubble.
The assembly of BTA factors in sequence, it need not be, but discerned by experiments, TFIID binds to TATAA site, how it is identified, it does so by TBP and TATA sequence specificity. It is not a chance but it is determined by certain proteins bound to their specific regulatory regions nearby which might have been identified during development.
Once components are placed in their respective sites, they can interact with upstream elements, whatever they are, whichever they may be, and by conformational changes in the protein complex, induce the opening of ds DNA into transcriptional bubble, which is required for ensuring transcription. Before initiation of transcription, chromatin has to be remodeled in such a way at least some prime transcriptional factors identify the said gene regulator elements and bind and induce changes in chromatin structure. There are complexes of remodeling proteins which are multisubunit structures.
SWI- is an yeast mating switching complex and contain ATPase units.
RSC –is Remolding Structure of Chromosome, a multisubunit components found in large numbers.
SNF -is a Sucrose non Fermentor associated with SWI.
BAH - components are proteins associated with bromodomain binding factors. Bromodomain consists of 110 a a sequence which binds to modified histone tails and recruits more HATS.
NURF - a nucleosome remodeling factor, it is a nucleosome stimulated ATPase consisting of subunits 215, 140, 55 and 38KD subunits.
ISWi -it is an imitation of SWI
RPD3- is a histone deacetylase. Some of the RNAP associated transcription complexes are called RPDs-
Core II- also contain mobile subunits -3, 10, 11 and a clamp 12 }
Histone acetyl complexes:
|
Type/Subunits |
Catalytic unit |
Domain type |
Target |
|
SAGA/15 |
Gcn5 |
Bromo |
H3, H2B |
|
PCAF/11 |
PCAF |
Bromo |
H3, H4 |
|
NuA3/3 |
Sas3 |
Neither |
H3 |
|
NuAA/6 |
Esa |
chromo |
H4 |
|
P300/CBP1 |
P300/CBP |
Bromo |
H2A,H2B,H3,H4 |
|
RSC (multisubunits) |
|
|
Histones |
|
SNF |
|
|
Histones |
|
SWI 1 to 5 |
|
|
Histones |
|
|
|
|
|
Yeast SWi/SNF (150 complexes per cell) = fly dSWI/SNF (brahma), =h SWI/SNF. Another similar complex called RSC is abundant and has 700 or more targets. Yeast iSWi ! and 2 = fly NURF, ChRC and ACF = HRSF,hACF/WCFR/hCHRAC,; there are others like NuRD and Mi-2.
Histone deacetylases
|
Type |
subunits |
Domain type |
target |
|
Sin3 complex/7 |
HDAC1/HDAC2 |
neither |
|
|
NuRD/9 |
HDAC1/HDAC2 |
chromo |
|
|
SIR complex/3 |
SiR |
neither |
|
|
RPD3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
Histone Methylases:
|
Type |
subunits |
Domain type |
target |
Type |
|
SuV39/CLR4 |
|
Chromo |
H3-Lys9 |
|
|
SET1 |
|
Neither |
H3-lys4 |
|
|
PRMT |
|
Neither |
H3-Arg3 |
|
|
|
|
|
|
|
DNA methylases attract histidine methylases, which may then get associated with Hp1 type of proteins. Some other proteins like RAP1 binds to methylated histones and recruits SIR proteins such as SIR 2, 3 and 4. SIR 2 is histone deacetylase.
Mediator complexes: This is a multisubunit complex consisting of 20 or more subunits, they are made up of some common proteins among the species and some are tissue specific; they stimulate basal transcription via activators or coactivators, they actually act between BTA components and coactivators or activators and or factors bound to enhancers or Upstream activators. They can also stimulate CTD phosphorylation.
Yeast MC:
Srb2,4,5,6 and 7, Med 1,2,6 and 7, galK, Pgd1, Nut1,2 and few others.
Metazoans MC-general:
CRSP, NAT, ARC, DRIP, TRAP, MED, PCC, SKB.
Chromosome remodeling during Gene Expression:
Regulation of gene expression in eukaryotes is more complex and intrinsic. The genome is organized into a nucleoprotein complex of different orders.
Chromosomes bear genes of different types such as protein coding; rRNA, tRNA and other RNAs such as RNAis, and they are expressed differentially during development and also in tissue specific manner during or after development. Even after development genes are expressed in tissues in response a variety of signals.
Chromosomes by themselves go through condensation and decondensation differentially during cell cycle and even after cell cycle.
Chromosomal changes has been observed during cell cycle, from relaxed at interphase or Go to highly condensed at metaphase. At interphase substantial number of genes is expressed in tissue specific manner. At metaphase the chromosome is condensed to such an extent all genes are shut off. Once the cell derivatives differentiate and develop into specific cell types, chromosomes relax get attached to the inner surface of the nuclear membrane proteins through their heterochromatin loci. Chromatin is also attached to nuclear matrix proteins, whose composition and nature is not yet clear. The relaxed euchromatin region is accessible transcriptional complex, it does not mean that all those genes present in euchromatic region are expressed, it is not so.
It is logical to expect that a chromosomal locus where gene are expressed, requires unwinding and the nucleosomal structures are relaxed and at least some part of the DNA of the said gene is free from histones for the binding of transcriptional complex and its related factors. Whether or not regions that are active in gene expression can be tested by DNAse1 treatment or micrococcal nuclease, which on partial digestion the DNA wherever it is free DNA is digested completely and wherever nucleosomes are found, only the linker DNA is digested. Nucleosome bound regions show ladders, the free region is free. This can be observed by gel electrophoresis. So the DNA of a gene is in active state should be free from histones, and mostly it is of promoter regions.
Gene expression at grand scale can be observed in cells of certain systems and of specific developmental stage.
1. All eukaryotic cells irrespective of species, do exhibit rRNA synthesis all the time in the nucleolar region of the nucleus. Requirement of rRNA to any cell is very high and so requires the transcription of rRNA all the time. To supply such high quantity, the DNA of rRNA genes (from the secondary constriction region) has looped out as nacked DNA consisting of 200 to 600 rRNA gene repeats. And one can visualize the each of the rRNA genes are in the process of transcription on the open DNA, and one can observe Christmas tree like transcripts arranged-from the nascent to old transcript. In this the chromosomal DNA is totally devoid of any histones, and the only proteins associated are RNAP I and its associated factors. This goes on 24hrs and day 365 days a year.
2. Another system that shows such grand scale transcription at a particular stage of development is Xenopus oocyte formation stage. During meiosis at pachytene- diplotene transitory stage, one finds meiotic chromosomes are maximally elongated and one can observe large number of granular structures all along the length of each paired chromonema threads. Some chromomeres in such synapsed chromatin are looped out into nacked DNA of 5kbp to 50kbp long. Such DNA is found to be actively transcribed. Such kind of transcription is required for the future development of egg into an embryo after fertilization. This is a preparatory stage where the developmental required proteins, rRNA, mRNAs and protein factors are produced and stored.
3. Another par excellent system is that of grand scale expression seen in insect larval development like drosophila and its related species. When the fertilized egg develops into larva, on reaching 11th day that is the entry into pupa stage. There is a dramatic transformation of the larval body into pupal structures and into insect perse. This huge transformation requires a grand scale expression all those genes involved in metamorphosis. The larval cells have four pairs of chromosomes and the total number of genes in the insect is about 18000 or so. The number of gene’ expression required for such transition has been assayed by micro array chips. But most of the genes exist as a pair of genes on their respective homologous chromosomes. But the requirement in such a short time is huge. In order to supply to such demands, each of the required genes are either to be expressed at phenomenal rates, which is short of expectation. Or each gene required has to be duplicated to thousand copies or so, so the expression of each gene suffices the demands. So insects have designed that their chromosomes in Salivary glands on 11th day undergo transformation in grand scale, visible under normal microscope, this the visualization of differential gene expression at cytological level. Chromosomal DNA undergoes rapid replication and chromatin duplicates itself into 1080 strands, it means all those genes are duplicated to 1080 copies. This means any of the se genes expressed; they are expressed thousand eighty times more. Starting from the early 11th day larva till the pupal initiation, sets of genes are expressed temporally, it means at the early stage one set of gene are expressed, as the development progresses, another set of genes start expressing and the early genes expression regresses. By the end of this progression thousands of individual genes are expressed and thousands become silent.
The polytene chromosome in its glory show 5000 or more very prominent chromosomal bands with 1080 duplicated threads (copies). Each of the bands open out into open DNA loops from 5kb to 50kbp size and their nacked DNA is associated with transcription machinery producing transcripts required to be translated and some to be stored. The opened regions look like puffs (chromosomal puffs), some of the long loops, identified by Balbiani are called Balbiani rings.
Such type expressions are also found in Phaseolus vulgaris embryo development at endosperm haustorial stages.
From the above description for gene expression the DNA has to be dissociated from histones, for they act as blocks for accessing transcriptional and its accessory components. But chromosomes are built out of four histones i.e. H2A, H2B, H3 and H4 and H1, they from and constitute structural components of chromosomes.
But chromosomes are also associated with thousands of nonhistones, which can be differentiated from histones from their very nature of being little acidic; not all of them. Most of them are involved in regulation of gene expression, DNA replication, DNA damage repair and maximum number of nonhistones found are to be Topoisomerases for they have to remove super coiled DNA into relaxed forms either at replication stage or at transcriptional stage. Along with Topoisomerases one notices the presence of gene activation and gene repression proteins all along the chromosomal threads, their loci depend on the cell type and stage at which they are found.
The number of such proteins involved in gene expression and repression requires a list of proteins, stage at which they express and where and how they act. Delineation of it requires the expertise of functional genomic professionals.
In this description, the expression and regulation of gene expression is dealt in two different functional aspects but inter-related structures and functions.
First Part:
This deals with how chromosomes are modulated either to open up for transcription or close and prevent transcription.
The role of Histones and Nonhistones in general:
It is very well documented that all cells in all tissues express certain set of genes for house keeping functions, they are called house keepings genes. Some are expressed in tissue specific manner; they are expressed using tissue specific factors. There are genes, any where in the organism, whatever tissue or cell types, are exposed to certain signal molecules, and signal specific genes are expressed.
In this context which of the chromosomal proteins prevent transcription and which allow transcription in non specific manner. To answer this question scientists have isolated DNA from an organism. They also isolated histones and nonhistones from different tissues.
An experiment is designed to show which of the chromosomal regions are involved in transcription and also in tissue specific manner.
Expt:
DNA isolated from a system is same; common DNA for the experiment.
Histones are isolated from specific tissues; Liver (L) and Brain (B); similarly non histones are isolated.
In one experiment nonhistones from Liver tissue is added to the DNA and allowed to express; experiments showed nonhistones expressed liver specific gene expression, along with common for both.
In the second experiment nonhistones from brain were added to the DNA, and they found brain tissue specific genes were expressed along with common for both.
Histones did not induce any gene expression.
Results: Nonhistones from brain expressed brain related genes and Liver specific nonhistones expressed liver specific Genes.
These results indicate that nonhistones from specific tissues have components to bind and express tissue specific genes along with house- keeping genes. But histones don’t induce any gene expression.
Second type of experiment:
Isolate a specific DNA segment; the add histones and then add RNA polymerase components, in this case rRNA DNA; the result-no transcription.
Use the rDNA and add RNA pol components first and histones later, transcription ensues.
Experiment tells us that histones prevent the initiation of transcription. In another experiment- histones were added first and RNA-Pol and TF-II D was added last, result no transcription. On the contrary IIF-D was added first and histones and the RNA pol complex was added later in succession. The gene is transcribed, it means, that TF-IID is the identifying factor of the promoter element, which Gene it might be.
Results: So the result indicates, that when DNA is free from histones the Transcriptional complex find their site and bind and initiate transcription.
In eukaryotes the promoter elements have more complex combination of sequence boxes to be recognized by specific factors.
The transcriptional activators bind to different element located at different sites from the START site. So also transcriptional repressors, do so by binding to specific sites identified by sequences. So activators activate specific gene transcription and repressors repress specific gene expression. There are others called silencers and insulators, which have their own specific roles to play.
The regulator proteins of one kind are activators, co-activators, mediator complexes and transcriptional complexes. They bind to different structural elements and activate the gene expression in specific. So also the gene repressors, they can be specific gene repressors, specific silencers or general repressors. In some cases the not just few loci are repressed from expression, the whole chromosomal genes are totally inactivated, as an extreme case, but it is prevalent in higher systems.
The structural and functional features of both gene activators and repressors have different motifs and specific functions in gene regulation.
In spite of the variety of components involved in regulating gene expression, the overall mechanism from yeast to man is more or less same.
Chromosomal remodeling:
Chromosomal activation:
At chromosomal level the factors involved in activation- are nucleosomal dissociation or nucleosomal remodeling factors that release the histone octamers or move the nucleosomal structures from one position to the other either way.
Chromosomal DNA in eukaryotes is linear and very long ranging from 10^6 to 10^8 bp. The DNA is associated with histone and nonhistones proteins, where histones participate as structurl components and non histone act as functional components either in activation or repression of genes.
Two tetramers of H3, H4 and H2A and H2B; each of them are made up of 3-helical secondary structures with loops in between. Interestingly each of the histones have N-terminal tails with specific sequence of amino acids, but among them H2A has c-terminal tail also. The helical motifs are called Histone folds and sequence of histone tails often referred to as histone code for they dictate the tail modifications at specific amino acid residues. The histone octamers is wrapped around by DNA and histone with their large number of positive charges bind to DNA at least at 14 points of contact. The length of DNA is approximately 146 to 150 bp. Such a structure forms a Nucleosome or Nu-body. Such octamers are organized in a series of successive bead like bodies, thus the chromosome in the first order of organization is called nucleosomal thread. The Nu bodies are regularly placed with DNA in between two successive Nucleosomes called linker DNA, and it is about 50-90 bp a little more. Histone 1 with its central globular and C and N tails binds to DNA found in the linker region in such a way, it occupies a cavity like region where DNA entry and exit around the histone is on the same side; it is at this site the H1 binds. H5 is a H1 variant, similarly some of the histone one finds in Centromere also differs to some extent, they are called CEN histones. When H1 binds to nucleosomal thread (11nm thick), it takes coiling called solenoid or it may exhibit compact zig-zag conformation as made out in electron micrograph; this is the second level of chromatin organization; this thread is 30nm thick
All histones have 3 helical domains as their histone folds, central long helix flanked on either side by smaller helices with loops in between.
The nu body is ~110A diameter and ~60A thick. The DNA wraps around the histone octamers by 1.65 turn in left-handed super coil, the left-handed super coil provides an unwinded state of DNA.
When chromatin duplicates at S-phase the replication fork drives through ds DNA separating parental strands into leading strand and the other as lagging strand. As the DNA strand separate the histone octamers distribute randomly and equally among the newly produced ds DNA. Histones that are synthesized organize into H3x2 and H4x2 tetramers and they bind to ds DNA and then H2Ax2 and H2Bx2 join. The H2A, H2B and H3 and H4 tetramers superpose in the formation of an octamer complex. The assembly is facilitated by acetylation of H3 histones and Nucleoplasmin and probably topoisomerases might be involved. In the formation of new chromatin threads nucleoplasmins play an important role as Chaperonins.
The third level organization of chromatin is the 30 nm fiber will be soon coated with various nonhistones protein, among them Topoisomerases and HMG proteins are found in large numbers. Histone depleted chromatin shows a central structural protein called scaffold protein and the DNA are found looped out of such structure, the loops are of various sizes 20 to 90kb (15 to 30um). A cross sectional view of a typical chromosome consists of a central scaffold protein from which nucleosomal threads loop out; the size of the loop is roughly about 0.3um and the central region is about 0.4 um, thus the thickness of the structure is 1um, it agrees with the thickness of the intact chromosome. A chromosome with 140 million base pairs could easily produce about 2000 such ~70kb sized loops. The DNA loops are attached to a protein complex with AT rich sequences called Matrix Attachment sites called MARS or one can call such regions as scaffold attachment regions (SARs). It is important to remember as and when the chromosomal DNA open out during replication and produce ds DNA, they get associated with their DNA binding proteins which were originally present before replication. The DNA binding proteins that bind provide site specificity and perhaps gene specificity, they can be activators, repressors or coactivators or any of them, but provide the identity of the gene. For ex. In yeast GAL 4 with GAL80 is bound to GAL1 promoter-activator region and represses the expression. Similarly gene activating factors and complexes perpetuate, so that the gene expression of the genes is expressed all the time.
Histone composition:
H1 215aa 23kd 1%R 29%K
H2A 129aa 14Kd 9 11%
H2B 125 13.8 6 16
H3 135 15.3 13 10
H4 102 11.3 14 11
H1 = 208 (215) aa, has central globular body with N and C terminal tails. H1 composition varies among the species. It has a 5 R s and 53 K s and 5 D& E s.
H2A = 129 aa, it contains both C (39 aa) and N terminal tails, with 12 R, 14K and 7 D/ Es.
H2A = N S G R G K Q G G K A R A K A K T R S S R A G L
H2B = 126(125) aa, it has N-terminal tail with 7R, 21K and 10 D n Es.
H2b =P E P S K S A P A P K K G S K K A I T K A Q K K D G K K R K R S R K.
H3 = 136 (135) aa, it N tail contains 18R, 13K and 11 D n Es.
N-A R T K Q T A R K S T G G K A P R K Q L A T K A A R K S A P A T G G V K K
H4 = 103(102) aa, N-tail contains 14R, 11K and 7 D n Es.
n-S G R G K G G K G L G K G G A K R H R K V L R D
Modification of H3 and H4 tails has an important effect on chromatin condensation and decondensation.
H3 = 1A-R-T-K-Q-T-A-R-K-S-T-G-G-K-A-P-R-K-Q-L, where K9 and K14 are acetylated in euchromatin, even S9 can be phosphorylated.

www.zoology.ubc.ca; imgarcade.com
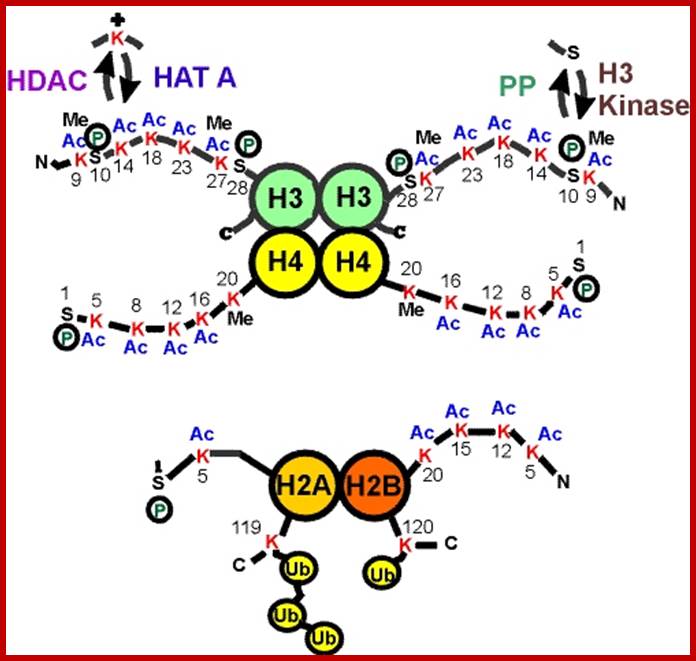
www.biomatics.org
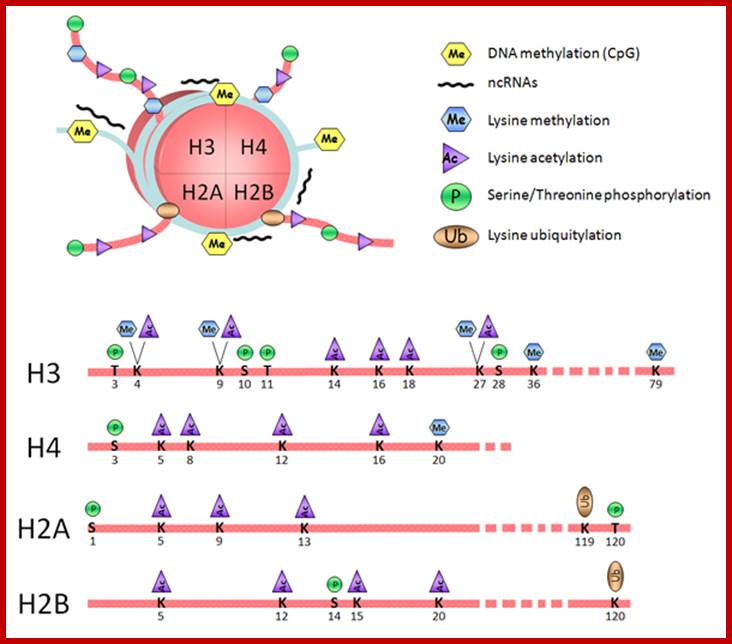
www.biochem.slu.edu
H4 = S-G-R-G-K-G-G-K-G-L-G-K-G-G-A-K-R-H-K, where R3 can be acetylated in euchromatin. The K can be acetylated or it can methylated, similarly the R can be methylated and serine can be phosphorylated. Specific modifications at specific sites have tremendous effects on chromatin activation (loosening) and chromatin inactivation (compaction).
acK = Acetylation to K (lysine,
meR = methylation to R (Arginine),
meK = methylation to K,
PS = phophorylation to serine,
Uk = ubiquitination to K,,
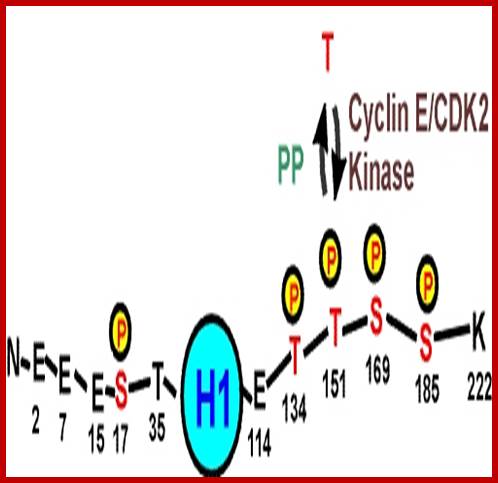
The order of amino acids in the H3 and H4 tails play significant positional role in chromosomal activation or inactivation (activation of a gene(s) or suppression of a gene(s)). David Allis proposed that histones N-terminal amino acid sequences provide encoded information, what is called Histone code, where certain modifications evoke certain chromatin-based functions, a combination of modifications evoke specific biological functions; here they can activate or inactivate a gene or gene functions.
Transcriptional activation via histone modification and remodeling:
David Allis concept of Histone code provides an input where active chromatin, means any genes in that region are expressed or expressing. Such chromatin shows its H3 is acetylated at K9, K14 and H4 K5 and its H3-K4 and H4 R3 are methylated. But condensed chromatin i.e. inactive is associated with H4-K12 acetylated, and H3’s k9 is methylated. Nucleosomes are also disposed for phophorylation at Serine10 and 28. The combination of modifications at specific amino acids in the N-tails H3 and H4 have intrinsic ability to change the shape of chromatin to open for transcription or close to transcription at specific loci or sites.
Histone modifications during early cell division and determination and differentiation provide those epigenetic markings assisted by the binding of specific factors to specific sites in the region provides the identity of the gene or genes to be expressed or not to be expressed in specific tissues or in specific environment or inducer or repressor signaling. This epigenetic inheritance of histones during determination and differentiation is yet be discerned beyond doubt. It is also provides markers for the association of gene specific or tissue specific factors or both, thus provides the inheritable constitutional input for the next generation of cells or tissues, which can respond to various inputs. To give a simple example of yeast GAL genes regulation; GAL1, GAL7 and GAL 10 are located nearby on the chromosome and their gene products are involved in utilization of Galactose in the absence of Glucose. The genes located at specific loci are bound by GAL4. The GAL4 binding remains at the location for any number of generations of cell lineages and remain the said genes repressed. GAL4 is actually, an activator, but GAL 80 by binding to GAL4 suppresses GAL4’ activators function This is an unicellular cell system, this may apply to multicellular system but indicates clearly that cell types generated during development contain specific factors associated with chromatin, which on stimulus can be expressed. Epigenetic is the hall mark gene regulation in Eukaryotes and Prokaryotes. In Prokaryotes, specific operons are repressod by specific repressor bound to their regulators and the repression perpetuate for many many generations. When an input is provided the repressors are removed and operons express. This is the one of the mode of gene regulation either in terms expression or gene repression. It is very important to remember that majority of the gene are expressed as house keeping genes, which are required as sine quo non components.
HATs:
It has been found that most of the genes expressed are found in chromatin loci where their specific histone tails are acetylated. And those genes remained suppressed or not activated are found to have methylated or deacetylated or the combination of both. This needs not be the case in all and exceptions are found.
HATs are components of multisubunit transcriptional coactivators:
Vincent Allfrey (in 1960s) discovered histone acetylation and deacetylation involved in transcriptional activation and inactivation in EuK (eukaryotic) systems. In 1990 HAT and DHATs proteins involved were identified and they found to act at specific positions of the Histone tails and some of them are well characterized. All HATs use acetyl-coA as group donors. As there a large umber of them they are grouped as five HAT families.
GNAT family: Gcn5 related N-acetyltransferase. GCN5 was first found in
Yeast; it is well characterized; it is related members are Gen5L, and PCAF ( p300/CBP –associated factors); p300 and CBP means cAMP response element binding element protein, they are homologous transactivational factors. Hat1 is found in cytoplasm and acetylase histones before they are transported into the nucleus.
MYST family: named after founding members- Moz, Ybf2/Sas3, and Sas2, and Tip60.
P300/CBP family: They are coactivators; when they are recruited to their PIC co activators or enhancer proteins, they acetylate histone tails in the vicinity where they are bound.
TAF Family; They are associated with TBP protein in RNA pol assembly complex. TAF1 (also called earlier as TAFII 250) is the largest protein of TFIID complex that binds as the first component in the assembly of PIC.
The SRC family: Steroid receptor co activators. They have a variety of functions in transcriptional activation or as silencers and also implicated in regulation of cell cycle and transcriptional regulation.
Most of the HATs act as multiple subunit complexes of 10-20 subunits, they form complexes such as SAGA (spt/Ada/Gen5L acetyl transferase), PCAF complex, STAGA (spt3/TAF/Gen5L acetyl transferase, where its HAT is Gen5L), ADA a transcriptional adaptor, TFIID –contain TAF1, TFTC (TBP free TAF-containing complex, NuA3 and NuA4 (Nucleosomal acetyl transferases of H3 and H4.
It is also interesting to find most of the above said complex does contain some common subunits such as Gcn5, Ada2 and Ada3 which are common to 14 subunit SAGA complex. Likewise, Tra1 is common for both SAGA and NuA4, Tra is a homolog of phosphoinositide 3-kinase; it interacts with specific transcriptional activators such as MYC. It is also a fact that several HATs contain one or more TAFs. For example, SAGA contains TAF5, 6,9,10 and 12. PCAF contains TAF 9, 10 and 12 but contain their close homologs PAF65b and 65a.
Look at the TAF6, 9 and 12 are the structural homologs of H3, H4 and H2b respectively. Thus, one can say they actually form an associated complex to interact with TBP.
Most of the HATs target their components to promoters and they found to be so in active genes. The complex of HATs and their specificity is more intrinsic and it is only now people started to understand the activation of specific gene or group of genes,
The million-dollar question what makes specific HATs identify which loci or genes to be acetylated or is it a general phenomenon? The answer lies in the type of HAT and the type of the site identified by certain factors, what are they?
Very interesting aspect of HAT associated transcriptional coactivators contain (mostly all) contain a ~110 residue module called a bromodomain. Any protein that contains this domain binds to Acetylated Lysine moiety of Histone tails.
For example, Gcn5 consists of a HAT domain followed by a Bromodomain. In the case of TAF1, it has N-terminal kinase domain followed by a HAT domain then two successive tandem Bromodomain. The TAF1 targets specific acetylated histone tails. It contains two nearly identical ant parallel 4 helix bindles, where one finds pockets to recognize acetylated tails and bind to them.
An example to illustrate the activity of a gene shows that the N-terminal tail of H4 contains K residues at 5, 8, 12 and 16; it has been found the acetylation of these said residues increases the transcriptional activity of the said gene,. So the question is what makes these specific sites to be acetylated among the million of nucleosomes and how the acetylases determine the sites to which bind are promoter elements or other activating sites.
It is also assumed that TAFs1 with its Bromodomain serves to target TFIID to the promoters that are found in the nucleosome containing sequence of a promoter or sequences in the vicinity of a promoter,
The critical feature is that the recruiting HAT-containing coactivators complex to a specific upstream element bound by a protein. Once such proteins bind to DNA, the HATs in them can promote acetylation of the nearing nucleosomal Histones and loosen the complex for the assembly of transcriptional complex to initiate transcription or to wait for other activators to act either to activate or suppress the activation.
In all probabilities chances of binding these acetylase complexes to specific sites or regions have to be identified another protein that is already bound to DNA as a site identifier. The logic is simple for all nucleosomes, irrespective the gene promoter they have in them, there should be an identity factor that is bound to DNA in sequence specific manner and such sites have to perpetuate during development. Without such markers, it is neigh impossible for the HATs to identify the sites.
Histone Deacetylases (HDACs):
Histone Deacetylases by removing acetyl group allow other components to bind or otherwise makes the region inaccessible for the assembly of transcriptional components including PIC or BTA and other upstream factors that regulated the gene expression, thus repress the expression a gene. Deacetylation of specific lysine otherwise of histone tail, provides an opportunity for other enzymes to modification of the tails in different ways. Example for it are methylases that methylate histone tail at specific amino acid residues, which need not be the same residue that is deacetylated. They can be different. Deacetylases provide an opportunity for other proteins to bind and modify in different ways, such as methylation and phophorylation or not.
Among the many, ten, HDAs have been identified for certainty. There are ten HDAs in yeast, 17 in humans. HDAs have a family of proteins such as Class I and Class 2 etc. There are at least three classes- such as ClassI, Class II and Class III; most of them are multisubunit complexes.
In humans the HDACs consist of the said three classes: Class I-contain HDAC 1, 2, 3 and 8. Class II consists of HDACs 4, to 7 and 10. And the Class III contains Sirutins (SIRT1-7) (SIR for Silent information regulators). Most of them are multisubunit complexes, where some are common to all the classes and some are specific to each of the classes. Some of the components are –Sin 3, NurD (nucleosome remodeling histone deacetylases), Co rest (co repressor of RE1 silencing transcription factor), NCOR (nuclear hormone receptor co repressor), SMRT (silencing mediator of retinoid and thyroid hormone receptor), Most of them serve as transcriptional co repressors at different sites and with different composition. For example REST on binding to its target, recruits CoRest and SIN3, which together repress the expression of the gene where they bind.
Histone methylation:,
Lysine and Arginine residues in H3 and H4 tails are the targets for methylation by Histone methyl transferases (HMTs). They use SAM as the methyl donor. The enzymes have SET domain [(Su (var) 3-9, E (Z), Trithorax], which have catalytic sites. Interestingly no demehtylases have been identified, which means methylation is not reversible (?). But in some cases, trimethylation of a histone tail is demethylated to di methylated sites.
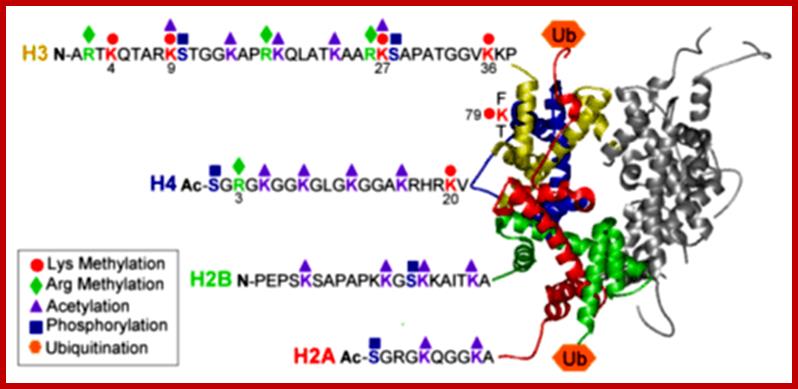
www.ag.perdue.edu
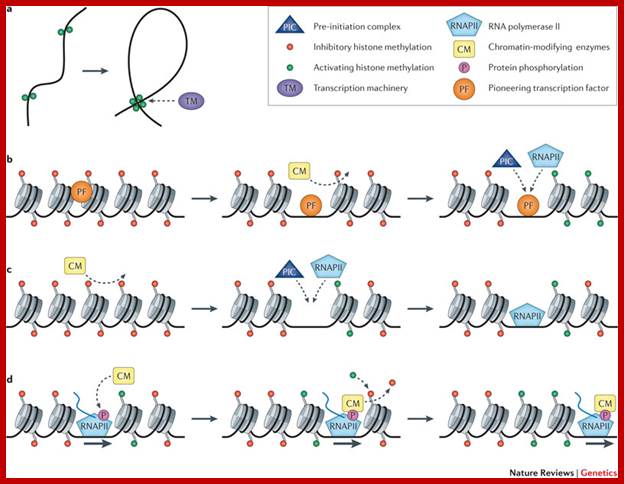
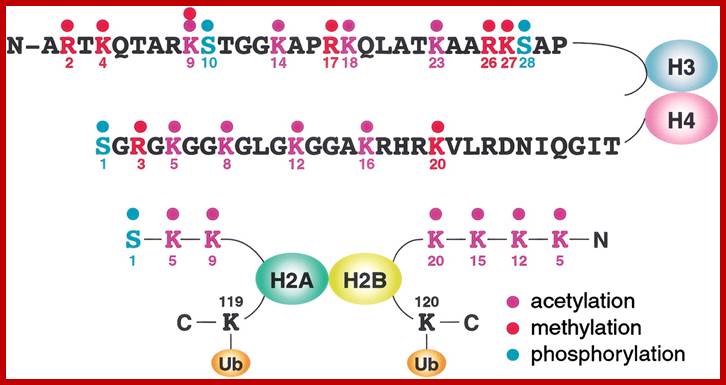
www.uniroma2.it
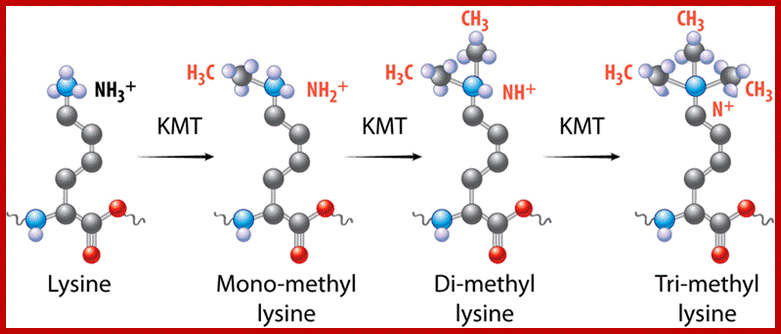
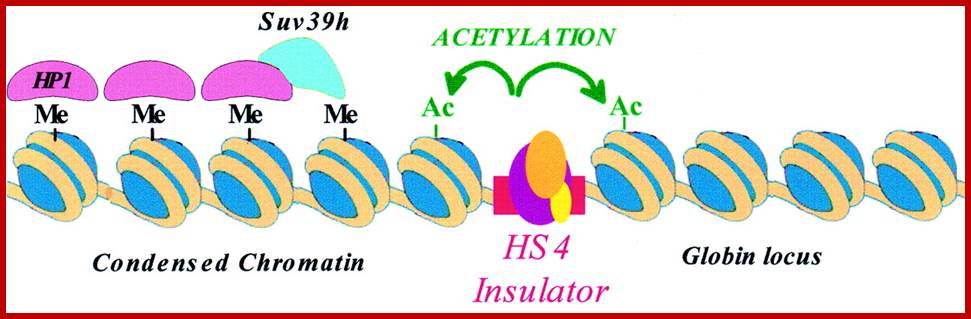
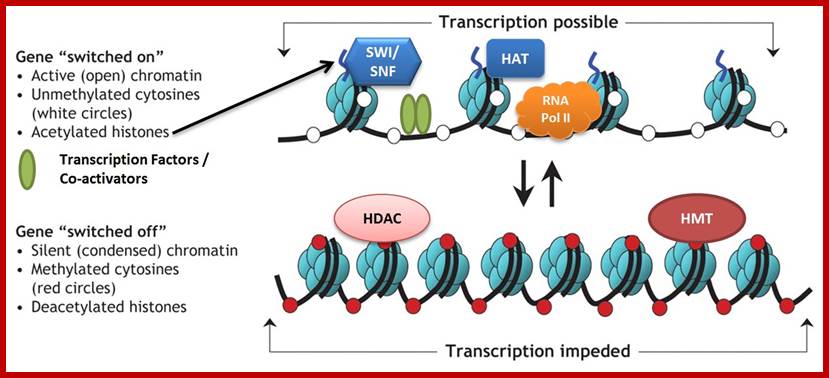
en.wikipedia.org
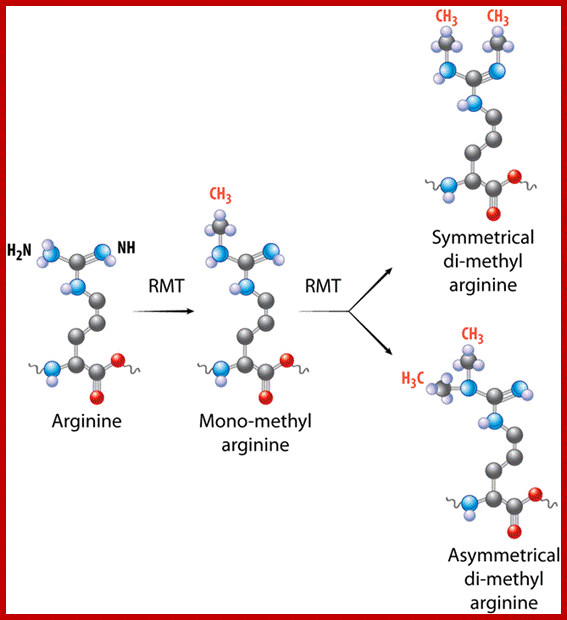
http://www.epizyme.com/epigenetics
Methylated histone tails are recognized by proteins containing chromodomains. For example, methylated H3 at lysine 9 is recognized by chromodomain containing Heterochromatin 1 (HP1) protein. The binding of HP1 recruits other proteins to control chromatin structure and gene expression. The said enzymes have active sites as clefts or grooves where only such tails of histones bind with specific amino acid (s) if methylated. This binding can lead to spreading of nearby chromatin to be silenced. This happens due to HP1 that is bound recruits HMT Suv 39h protein complex that methylates the neighboring histone tails, which recruits More HP1 proteins. Thus, heterochromatin spreads. However, the spreading heterochromatization is often checked by insulators found in the pathway of heterochromatization. Ex – chick b-globin clusters recruit HATs that acetylates H3 lys9 at nearby nucleosmes, thus it checks the spreading of heterochromatization. Question is what makes the methylation mediated HP1 binding leads to heterochromatization or what we call it as condensation of chromatin?
Histone ubiquitination and Transcription:
Ubiquitination to protein via lysine ligation leads to proteosome mediated protein degradation. But in yeast ubiquitination of H2B Lys 123 mediated by ubiquitin ligase, Rad6 and E3 Bre1 are responsible for methylation of H3 K4 and k79. This modification results in silencing genes located near telomere. It is suggested that H2B ubiquitination (not for destruction) functions as master switch that controls site selective methylation and silencing telomeric gene silencing. It is interesting to find that TAF1 subunit of TF IID is involved in post translational modification of has been found in Drosophila. It appears ubiquitination of certain Histones regulatory sites emerging as regulators.
Chromatin remodeling complexes:
Chromatin is complexed DNA-histones as nucleosomes superimposed by nonhistones. Such a thread is often exists as condensed and relaxed threads. It is believed that relaxed region is more open for transcription than the condensed ones. Remodeling means the chromatin condensed to be decondensed or decondensed to condensed; it is done by chromatin remodeling proteins.
This remodeling of chromatin is performed by certain multi-subunit protein complexes that are associated with chromatin. It is also important to remember that there are several thousands of proteins, called site specific identifier proteins, bound to chromosomal DNA even though they are in the form of nucleosomal threads in various states. Some such bound proteins are perpetuated and some may be added with time and state of the cell. Multisubunit complexes, which are ATP dependent, provide fluidity to chromatin. The chromatin is also associated with chromosome remodeling multi subunit protein complexes. Most of them ATP driven, so they remodel the nucleosmes to move from one site and down stream so a region containing promoter is made available for regulatory proteins to bind and act.
One such complex is SWI/SNF, they were discovered in yeast and thay make HO genes to express. (SWI means switching defective-mating genes). And SNF means Sucrose Non Fermentor. The SWI/SNF complex is 110KD consists of 11 subunits. It is able to express 3% of the yeast genes.
Another complex called RSC (Remodel the Structure of Chromosome) is 100 times more abundant than SWI/SNF and they share two subunits of SWI/SNF. RSC contains hmologs of SWI/SNF, called as Sth1, they are called as swi2/snf2 in SWI/SNF. Swi2/snf2 are ATPases and two of the subunits of RSC have bromodomains so they bind to acetylated Histone tails.
The remodeling complexes are grouped-
1. SWI/SNF complexes (include ATPases sw2/snf2, RSC, Brahma BRM (Brahma related protein) BRG1-Brahma related gene 1.

www.oncologydiscovery.com
2. ISWI- for imitation switch components, ACFs-ATP utilizing chromatin assemble and remodeling factors, CHRAC-chromatin accessibility complex. NURF- Nucleosome remodeling factor and NSF-Nucleosome remodeling and spacing factor.
3. Mi2 complexes –they include human NURD also contain HDAC1 and HDAC2.
4. Many of these complexes contain bromodomain or chromodomain, when they bind they recruit other factors to their target.
www.Intechopen.com
It is known that some these factors are associated or bound to transcriptional activators. Having ATPases factors use ATP energy to distort binding of DNA to Histone and push the histone complexes along the DNA like helicases, they free the DNA from nucleosomal structure, so a segment of DNA is left free for the assembly of Transcription factors and their associated components. Gene activation starting from identifying the loci, assembly of various factors such as chromosome remodeling factors, assembly of PIC or BTA, Mediator complexes, TFs, architectural factors, co activators, co repressors, even some enhanceosomes, histone and chromatin modifying components and many more are invovlved.