Cell Cycle2
Cell Cycle and Cell Cycle Genes:
Cell with all its metabolic biochemicals and the genome, goes through cell division, to increase the number of cells when required. The process is regulated by a host of factors and genes, some promote and some monitor and check every step in during cell division whether it is correct or not; if no mistakes are found then the cell moves to the next stage and completes its cell cycle. Estimates indicate at least 800 genes control cell cycle (Swetha et al).
https://en.wikipedia.org
https://en.wikipedia.org

https://en.wikipedia.org
Cell cycle check points- Restriction point, G1-S heck point, M-Spindle check point, Post replication checkpoint
Cell cycle control molecules were first discovered through cell fusion experiments in the 1970s. The fusion of cells in different stages of the cell cycle (to form a heterokaryon) demonstrated that latter stages possess factor trigger progression. Principles of Cell Biology; http://www.mun.ca/biology
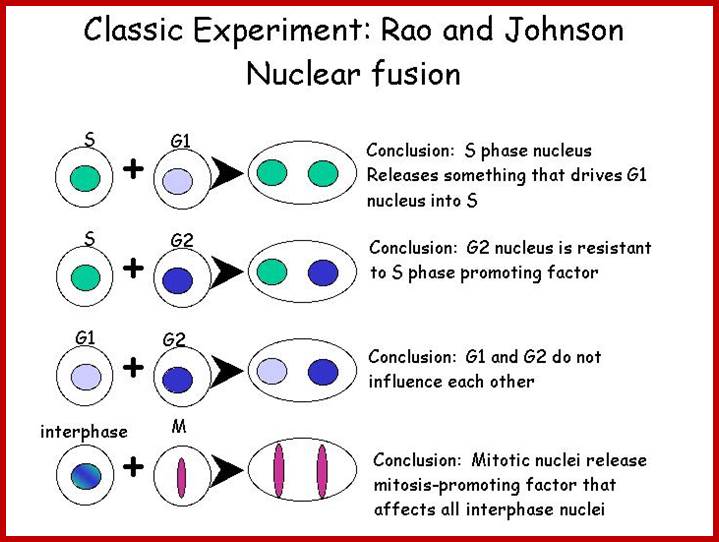
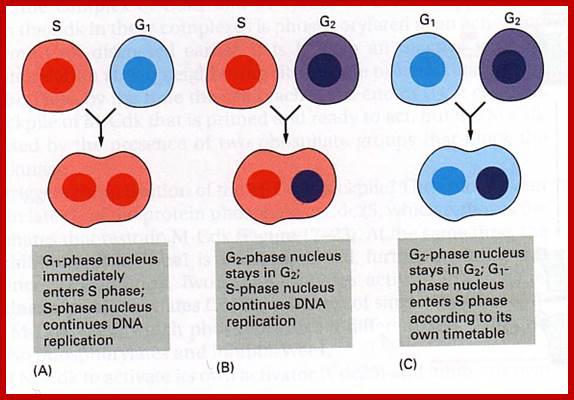
www.personalpages.manchester.ac.uk In classical experiments as shown above certain molecular events during each of the stages generate a set of factors and they are responsible for executing the stage and perhaps provide signals for the next stage. For example, when a cell in S-stage is fused with G1 stage, the cell in G1 stage is stimulated to proceed into S-phase. But if a cell in S-stage is fused with a cell at G2 stage, nothing happens, which means the components found in S-phase cells have no effect on G2, because the cells at G2 cells have already achieved what the S-phase components can provide. Fusion between G1 and G2 does not result in any changes in each of them. But if an Interphase cell is fused with a cell at M stage the Interphase cells directly enter into M-phase with disastrous consequences. The Interphase cell is not yet competent to enter into M phase, but M phase cells have all the components for chromosomal separation. So there is regulation at each of the entry points called check points, which is tightly regulated.
In classical experiments as shown above certain molecular events during each of the stages generate a set of factors and they are responsible for executing the stage and perhaps provide signals for the next stage. For example, when a cell in S-stage is fused with G1 stage, the cell in G1 stage is stimulated to proceed into S-phase. But if a cell in S-stage is fused with a cell at G2 stage, nothing happens, which means the components found in S-phase cells have no effect on G2, because the cells at G2 cells have already achieved what the S-phase components can provide. Fusion between G1 and G2 does not result in any changes in each of them. But if an Interphase cell is fused with a cell at M stage the Interphase cells directly enter into M-phase with disastrous consequences. The Interphase cell is not yet competent to enter into M phase, but M phase cells have all the components for chromosomal separation. So, there is regulation at each of the entry points called check points, which is tightly regulated.
Restriction point: Before the Restriction-point, the cell requires these extracellular stimulants to begin progressing through the first three sub-phases of G1(competence, entry G1a, progression G1b). After G1b external signals/ stimulation not required; extra cellular signaling must be maintained, and the cell must also have access to sufficient nutrient supplies to support rapid protein synthesis. Accumulation of cyclin D's are essential; cyclin D acts as a mitogenic signal sensor; Active cyclin D-Cdk complexes phosphorylate Retinoblastoma protein (pRb) in the nucleus. Unphosphorylated Rb acts as an inhibitor of G1 by preventing E2F-mediated transcription. Once phosphorylated, E2F activates the transcription of cyclins E and A. Active cyclin E-cdk begins to accumulate and completes pRb phosphorylation, as shown in the figure. Ling Chong You and Joe Nevins groups at Duke University in 2008 demonstrated that the bistable hysteric E2F switch underlies the restriction point. E2F promotes its own activation, and also promotes the inhibition of its own inhibitor (pRb), forming two feedback loops (among others) that are important in establishing bistable systems.
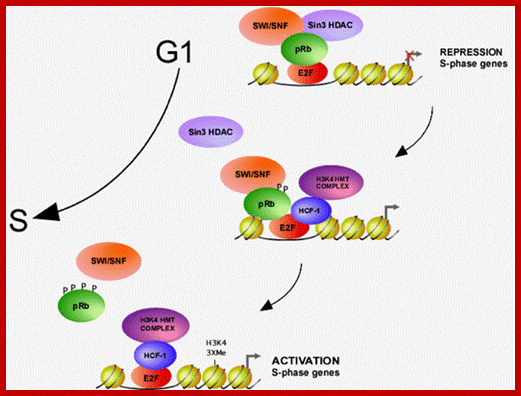
Proposed model for activation of S-phase promoters by E2F1; During G1/S phase transition, HCF-1 and its associated H3K4 HMTs are recruited to E2F-responsive promoters, replacing the pRb repressor complex. The E2F-HCF-H3K4HMT complex then activates transcription of S-phase genes. http://www.cdfd.org.in/
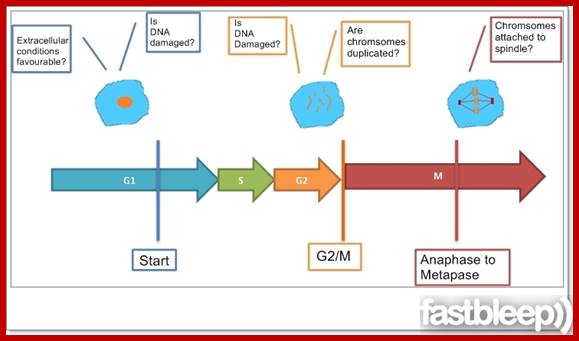
Checking the Requirements for each of the cell cycle phases; https://www.fastbleep.com
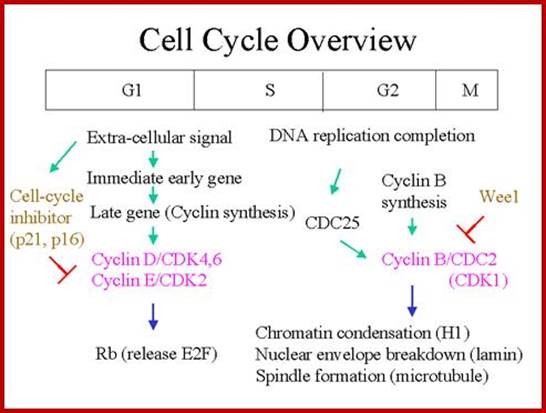
http://www.netcategory.net/
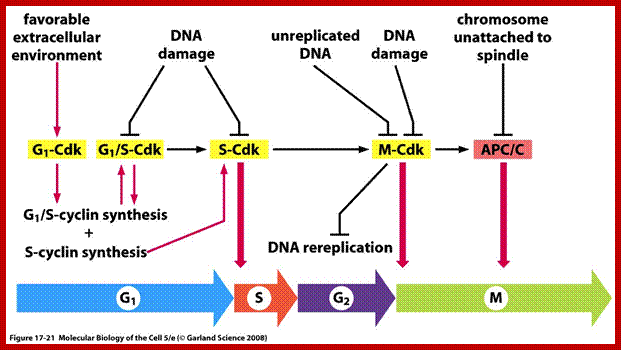
Factors that favour leading to the next stage; CDK and Cyclins are important, and there are factors that check the progress at each stages; https://www.studyblue.com
Krebs cycle meets the cell cycle; Role of mitochondria and the G1-S transition:
G1/S Checkpoint checks the existence of all conditions (nutrients and enzymes) required for DNA synthesis. Growth factors, hormones etc simply are signals supplying information about the local conditions (amino acids, glucose, NAPH, O2 etc.).

http://www.pnas.org; ;http://flipper.diff.org/
Eukaryotic mitochondria resulted from symbiotic incorporation of α-proteobacteria into ancient archaea species. During evolution, mitochondria lost most of the prokaryotic bacterial genes and only conserved a small fraction including those encoding 13 proteins of the respiratory chain. In this process, many functions were transferred to the host cells, but mitochondria gained a central role in the regulation of cell proliferation and apoptosis, and in the modulation of metabolism; accordingly, defective organelles contribute to cell transformation and cancer, diabetes, and neurodegenerative diseases. Most cell and transcriptional effects of mitochondria depend on the modulation of respiratory rate and on the production of hydrogen peroxide released into the cytosol. The mitochondrial oxidative rate has to remain depressed for cell proliferation; even in the presence of O2, energy is preferentially obtained from increased glycolysis (Warburg effect). In response to stress signals, traffic of pro- and antiapoptotic mitochondrial proteins in the intermembrane space (B-cell lymphoma-extra-large, Bcl-2-associated death promoter, Bcl-2 associated X-protein and cytochrome c) is modulated by the redox condition determined by mitochondrial O2utilization and mitochondrial nitric oxide metabolism. In this article, we highlight the traffic of the different canonical signaling pathways to mitochondria and the contributions of organelles to redox regulation of kinases. Finally, we analyze the dynamics of the mitochondrial population in cell cycle and apoptosis.
Mitochondrial Regulation of Cell Cycle and Proliferation;
A
putative model for the role of mitochondria in the G1–S transition; throughout
most of the cell cycle, mitochondria appear as a combination of either tubular
or fragmented morphologies. Surprisingly, at the G1–S transition, the
mitochondria coalesce into a giant, single tubular network. This network is
electrically coupled and exhibits a hyperpolarized mitochondrial membrane
potential (ψm). Because ψm is the ionic gradient used to generate ATP, it is not surprising that this unique
morphological and bioenergetic mitochondrial network appears to allow for
increased ATP generation. Based on ref. 1, and previous
studies in mammalian cells and lower organisms, the absence of this energetic
boost may trigger a G1–S checkpoint that involves the sequential activation of AMPK and p53 and ultimately the
down-regulation of cyclin E levels. In addition, ref. 1 suggests that increased
mitochondrial activity can positively regulate cyclin E levels and trigger
S-phase progression. Note, in this putative model, p53 regulates events in
several different contexts, including the transcriptional induction of p21, the
cell cycle regulator, and SCO2, a factor
that has been demonstrated to regulate mitochondrial oxygen consumption.
Antioxid. Redox Signal. 16, 1150–1180. Valeria Gabriela Antico Arciuch et al; https://www.ncbi.nlm.nih.gov
Cell Cycle Check Points:
Even before check point proteins operate the cell cycle is partially shows restriction points. The major restriction point lies in between G1 and S phase and check points between G2 and M-phase and another check point exists in the late M-phase at anaphase. DNA damage is critical for cells to divide, if any damage occurs it introduces a checkpoint, where until the damage is repaired, cell does not enter M-phase, this can happen at S-phase or at G2 phase; if the damage is beyond repair the cell is signaled for Apoptosis.
Checkpoints act at transition points where all the earlier events have to be completed before it progresses to the next stage. They also act as surveillance systems. It is a regulatory loop where initiation of one event depends on the completion of the earlier event, so progression through a checkpoint is strictly controlled.
There are three major check pints; G1, G2 and M; G1checkpoint is at the end of G1. Here, the cell evaluates whether the cell has all their inputs or not, to continue with the division process. The G2 is a very important checkpoint for the cell. At this time, the cell must evaluate if it has properly duplicated all of its chromosomes. If not, it may attempt to repair then move on to the next stage or it may simply abort it or commit suicide -Apoptosis. The third and the last major checkpoint occur during M phase. At this checkpoint the cell evaluates whether the tractile fibers of spindle apparatus have properly attached itself to each of the chromosomes at centromeric sites, and whether the rest of the cell is ready for cytokinesis or physical cell division. If something is wrong at this stage, the cell will often simply commit apoptosis. http://moodle2.rockyview.ab.ca/
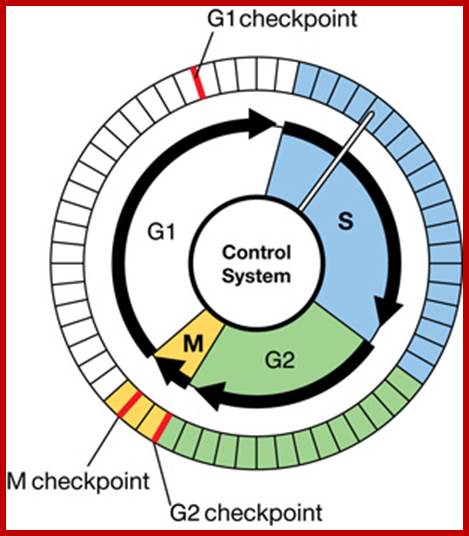
Three main check points;
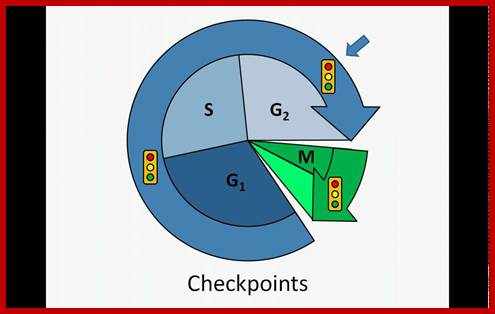
G1-S check point, G2-M check point and spindle check point;
www.youtube.com
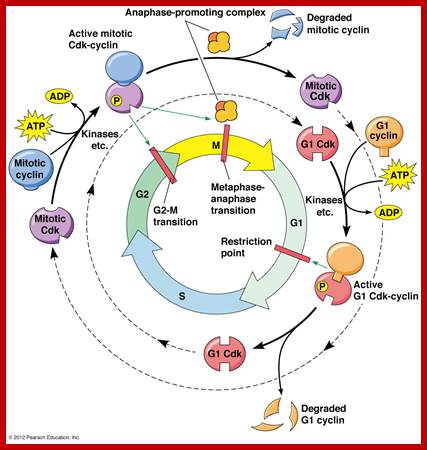
Stage specific CDK: cyclin and APC are involved in checking or progression of cell cycle stage by stage, step by step; ; www.mun.ca
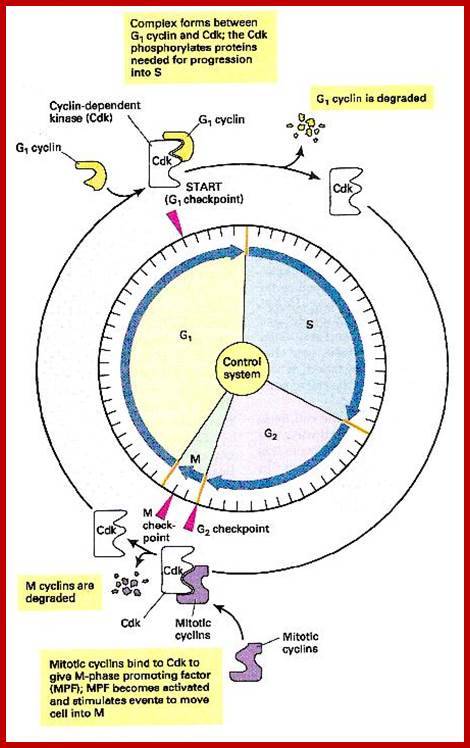
Stage specific cyclins once their function is completed they are degraded by proteasomal manner; http://www.fattuesdayproductions.com/
- Among the many cellular components involved, cyclin dependent kinases (Cdks) play a significant role. Cells employ more than 1000 kinases and also employ equal number of phosphatases. The beauty of the interplay of these two components is that many proteins and other cellular components rendered active when they are phosphorylated at specific stage and specific sites on them. In some cases, phosphorylation of certain components including proteins and other components inactivate them; depending upon the individual components, they become active when they are dephosphorylated. Thus, specific kinases phosphorylate specific proteins substrates at specific sites; thereby they activate or inactivate cellular components. Phosphatases in turn remove phosphate groups from specific sites in specific protein, so the substrate may be rendered active or inactive. Similarly, there are a host of inhibitors especially kinase inhibitors, and they play a pivotal role.
- In all this, the entry of cells into S phase is very crucial for the cell to divide and generate two individual cells with the genetic material equally duplicated and equally distributed without making even a single mistake; it is very very important.
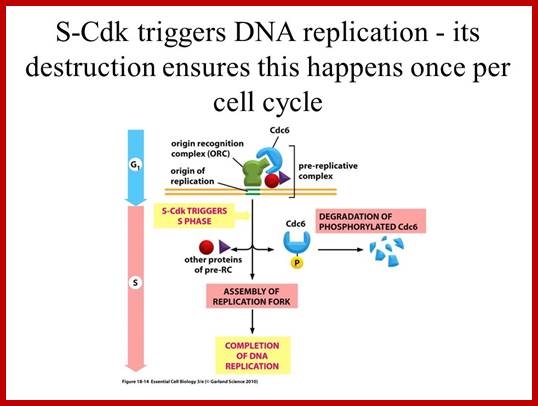
- Thus, DNA replication at S phase is critical. For initiating DNA replication exactly at S-phase and completion of it in S phase requires an input of many qualitatively different factors that have to be provided just before the start if replication. So, DNA entry into replication mode, completion of replication and separation replicated daughter DNA molecules in all its glory is controlled by the inputs of several factors. The diagram below shows some of the crucial components required and events that rigger the initiation of Replication at specific sites and at specific time is depicted.
- The S-phase depends on previous M-phase; till it is over another round of DNA replication won’t be initiated
|
|

Model illustrating general aspects of CDK regulation; CDK activation requires cyclin (CYC) expression and association. Cyclin/CDK complexes are kept inactive through association with CDK-inhibitory proteins (CKIs) and inhibitory phosphorylation by Wee1/Myt1 kinases (black circles). Activation requires ubiquitin-dependent proteolysis of the CKI, phosphorylation of the CDK by a CDK-activating kinase (CAK; red circle), and removal of the inhibitory phosphates by a Cdc25 phosphatase. Cyclin destruction leads to inactivation. Ubiquitin-dependent proteolysis of cell cycle regulators in late G1 and S involves cull in-based E3 ligases such as SCF, while in M phase and early G1 the anaphase-promoting complex (APC) is active. The exclamation figure denotes the active kinase complex, the large arrow indicates time; www.wormbook.org

Interplay between SCF and APC/C complexes. (A) During G1, APC/CCdh1 ubiquitinates Skp2, contributing to the post-mitotic down-modulation of SCF. (B) At G1/S boundary, SCFSkp2 activity increases, and CKIs substrates are ubiquitinated and degraded. This, in turn, activates cyclin/CDKs complexes that phosphorylate Cdh1, contributing to the down-modulation of the APC/C complex activity. (C) At the onset of mitosis, SCFβ-TRCP induces the degradation of the APC/CCdc20 inhibitor Emi1, increasing APC/C activity. Http://cardiovascres.oxfordjournals.org/
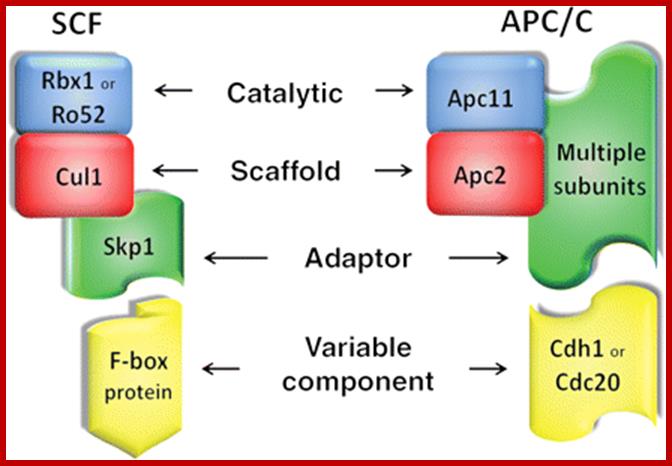
Structural similarities between SCF and APC/C E3 Ub-ligase complexes; Both complexes are composed of a catalytic RING protein (blue), a scaffold protein (red), and an adaptor protein (green). Variable components (yellow) give substrate specificity to the complexes: about 70 F-box proteins have been identified in humans, whereas APC/C is modulated by Cdh1 or Cdc20. http://cardiovascres.oxfordjournals.org/
Cycle of Cyclin synthesis and degradation:

Cell Cycle Regulation and check Points; WWW.IMM.MED.NCKU.EDU.TW;
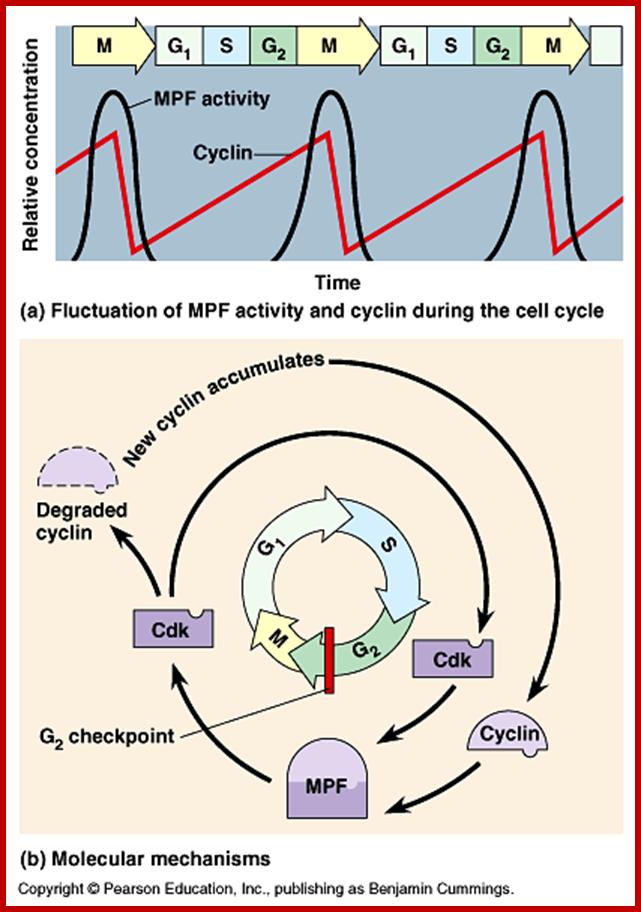
The figure above explains the linkage between cyclins and CDKs in the case of MPF and shows how the cyclin-Cdk complex induces passage into the M phase from the G2 phase; http://sharonap-cellrepro-p2.wikispaces.com/ www.mun.ca
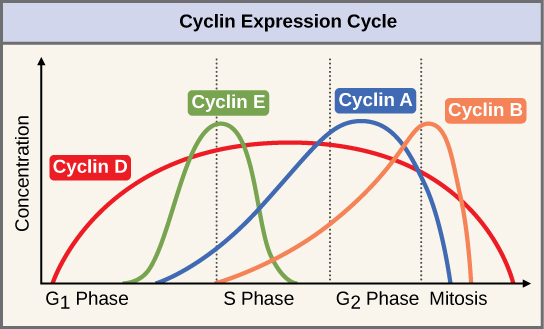
The concentrations of cyclin proteins change throughout the cell cycle. There is a direct correlation between cyclin accumulation and the three major cell cycle checkpoints. Also note the sharp decline of cyclin levels following each checkpoint (the transition between phases of the cell cycle), as cyclin is degraded by cytoplasmic enzymes. (credit: modification of work by "WikiMiMa"/Wikimedia (Commons), www.cnx.org; www.wiki.org.
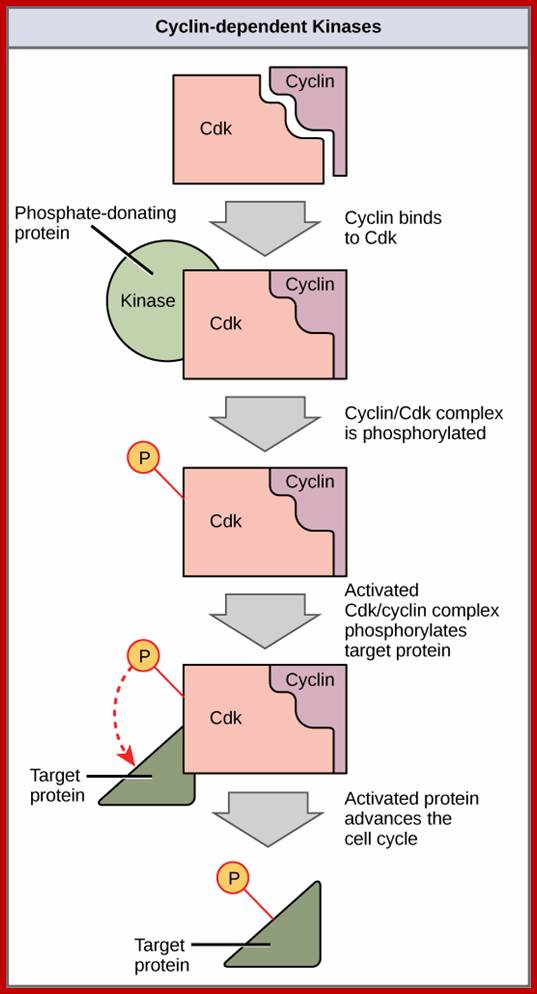
Cyclin-dependent kinases (Cdks) are protein kinases that, when fully activated, can phosphorylate and thus activate other proteins that advance the cell cycle past a checkpoint. To become fully activated, a Cdk must bind to a cyclin protein and then be phosphorylated by another Kinase. www. http://cnx.org/
The G1 stage, as said earlier, is the stage where cell prepares for DNA replication. The cyclins required at this stage are cyclins D. The required components for DNA replication, besides a large number of nucleotides and histone pools and DNA replication machinery, such as DNA polymerase, helicases, SSBs, and many other factors are required. It is during this stage or at the end of this stage transcription of genes required for DNA replication is activated. Transcription of the said genes requires transcription factors and their activation is sine quo non for the entry of the G1 to S-phase. If there is any damaged DNA at G1 stage entry into S-phase is prevented by the mediation of p53 and its associated components. If and only if all the required components for replication are provided then cell enters into S-phase.
- The S-phase is critical for the single stranded chromosome becomes double stranded by means of DNA replication, yet they are held together all along the length of the chromosome and also at centromeric region which has an elaborate structural organization called kinetochore (Centrosome). Each of the chromosomes contain one long dsDNA compacted by nucleosomal organization. Initiation of replication is initiated at specific sites called replication origin and it is governed by several factors. The number of Ori sites vary from one chromosome to the other based on the length of DNA in the chromosomes. Firing of replication is critical and it takes place only once in one cell cycle and second initiation is prevented before the M-phase is completed. During replication if there are any errors; they are fixed, and if the damage is beyond repair, the cell is subjected to Apoptosis. The progression of S-phase requires cyclin-E. Once their function is over these cyclins are degraded by SCF mediated process. The SCF complex consists of Cdc 53, SKP1 and Cdc4, all together targets Sic-1 the inhibitor of S-phase Cdk-cyclins. By ubiquitination process the Sic1 is degraded, this releases the Cdk-cyclin (G1) complex from inhibition.
http://slideplayer.com/
The G2 stage is again a preparatory stage for M-phase, which requires a whole set of proteins and organization of cellular components for chromosomal separation and cytoplasm division. If the DNA damage is not repaired in the S-phase and even in G2 phase the cell won’t enter into M-phase. Cells have in-built sensory system.
- The M phase, though short in duration, involves a major physical changes,; dismemberment left nuclear membrane and pore-complexes, dismemberment of endoplasmic reticulum, disassembly of microtubules and actin filaments to their respective subunits tubulins and actins, organization mitotic apparatus, including tractile fibers at centrosome, separation of chromatids and centromere, movement of chromatids to their respective poles and finally cytoplsmic division. These events can be visualized. Among the M-phase events the regulatory complex that plays an important role is ‘Anaphase Promoting Complex’ (APC).
Mutations of certain components that exert cell cycle control in yeast are: Cdc 28 at G1-à S; Cdc 28 at S phase; Cdc24 at G-2 and Cdc 34 M-à G1
Genetic analysis of single cell systems such as Saccharomyces cerevisiae (budding yeast) and Saccharomyces pombe (fission yeast) has yielded a wealth of database information. Combined with genetic data, Proteome search has provided information about the number of genes and gene products involved in cell cycle control. The database search to 95% accuracy shows that Homo sapiens have 99 gene products [28-protein for G1/S, 28 proteins for -G2/M, 23 proteins for -M, 41-Sphase, 24-others], Mus musculus contain 68 gene products and S.cerevisiae 87 to name only few. Important factors that operate cell cycle control system are cell cycle specific kinase complexes and few cell cycle kinase inhibitors. Among many of the kinases cyclin dependent kinases (CDK) are important; for their activity cyclins are required, hence the name cyclin dependent kinases. Kinases are effector molecules and cyclins are required for kinase effector function. Perhaps the first such kinase discovered was from yeast and it was called Maturation promoting factor (MPF), later it turned out to be Mitotic Promoting Factor (MPF) or mitosis Promoting Kinase (MPK). This protein complex turned out to be a serine-threonine protein kinase but dependent on specific cyclin. From a variety of sources many such cyclin dependent kinases and cyclins have been discovered their nomenclature has not been strictly adhered to international nomenclature rules, so there is confusion in identifying, which is which.
Cyclin dependent kinases (CDKs):
There are several CDKs, at least 11 or more. In general’ CDKs have a molecular weight of 34 kda, and they are monomer and function as kinase subunit i.e. as effector domain. It consists of N-terminal beta-sheet containing ATP binding site and an alpha helix with PSTAIRE sequence. The C-terminal has helical domain. When Cyclin binds the PSTAIRE region fits into Cyclin structure. CDKs are constitutively synthesized and found in reasonably higher concentration. When cyclin is not bound to CDK, the C-terminal loop can fold back and mask the ATP binding site and block access to protein kinase site. CDK is a protein kinase and responsible for phosphorylating several target proteins, thus activate several components that leads to the progression of M-phase and S-phase.
The N-terminal region contains phosphorylating sites at threonine 14 or at tyrosine 15; this depends upon the organism. These sites are adjacent to the substrate-binding site. Another site for phosphorylation is Threonine 160 in (CDK2) and Threonine 161 in CDC2 (it is also called CDK) from Schizosaccharomycs pombe.
Cyclins:
Cyclins are cyclin dependent protein kinase activators. There are several cyclins at least 30, which are synthesized and onces their function is over they are degraded (by ubiquitination and proteosome mediated process) in stage specific manner. The molecular weight ranges from 35 to 90 KD. Cyclins are made up of helices into which CDK snugs in. The 100aa long five-helix domain called cyclin box (shared by all cyclins). The C-terminal contains sequence of 9 aa, called destruction boxes, which is recognized by ubiquitination enzyme complex. However cyclins-C, F, G and H have structural relationship but not ll are involved in cell cycle regulation. Example cyclin-H/Cdk 7 dimers are associated with eukaryotic TFII-H.
Cyclin D; https://en.wikipedia.org/wiki
Cyclin destruction box:
Cyclin-A: RTVLGVIGD,
Cyclin-B: RTVLGVIGN,
Cyclin-B2: RAVLGVIGN.
Ubiquitination by E1, E2, and E3 target mitotic cyclins of anaphase Promoting Complex (APC) at the end of anaphase
CDK activity:
CDK is cyclin dependent kinase, its activity is regulated. The CDK has at its N-end has a Threonine 14 or Tyrosine 15 adjacent to kinase site for phosphorylation i.e. Thr 160 or Thr 161. If these hydroxyl amino acids are phosphorylated (by Wee 1); and Wee 1 is active when it is unphosphorylated and become inactive, if it is phosphorylated by nim 1 enzyme, the enzyme remains inactive; it will be active only when cyclin is bound (binding of cyclin opens up Thr160 site) and unphosphorylated Thr 14 or Tyr 15, but CDKs has to be phosphorylated at Thr160 or Thr 161. Dephosphorylation performed by activated cdc25 ( a phosphatase enzyme, becomes active when it is phosphorylated otherwise inactive). Phosphorylation of Thr 160 (161) is a must. This phosphorylation is believed to be by the enzyme called Cdk-activating kinase (CAK). Phosphorylation of Thr160 (161) can also be achieved by autophosphorylation once the CDK is active. The Cdk-cyclin dimmer protein is a serine and tyrosine protein kinase.
Cyclin dependent Kinase 6; CDK6: https://en.wikipedia.org/wiki/

The diagram representation of 3-D ribbon model of Cyclin-A and Cdk2 proteins; lmb.bioch.ox.ac.uk
A list of CDKs and Cyclins:
G1/S Regulatory Components:
|
|
CDKs |
Cyclins |
|
Mammalian & Frog |
Cdk2, 4 |
Cyclins D1, D2, D3, E |
|
S.cerevisiae (budding) |
Cdc 28 |
Clb B-1-4 (B-like) |
|
S.pombe (fission) |
Cdc 2 |
Cdc13 (B-like) |
G2 / M Regulatory Components:
|
|
Cdks |
Cyclins |
|
Mammals/frogs |
Cdk2, (cdc2,) |
Cyclin A, B1, B2 |
|
S.cerevisiae |
Cdk 28 (cdc28) |
Cyclin 1-4 (B-like) |
|
S.pombe |
Cdk2 (cdc2) |
Cyclin13 (cdc13) (A-like) |
*** Paul Nurse (UK), Thomas Hunt (UK) and Leland Hartman (USA) were awarded Nobel Prize for their work on Cdc cyclins.
Note that the cdc2 (cdk2) of mammalian system is equivalent to cdc28 (Cdk 28) of S. cerevisiae, which is equivalent to Cdc 2 (Cdk 2) of S. pombe. The Cdk term is used because each of them acts as Cyclin Dependent Kinase. The Cdk has a molecular weight of 34 KD. CAKs are CDK-activating kinases called CAKs.
Cdc 13 is a homolog of cyclin-B.
Combination of Cyclin-Cdk; Their Functions at Different Stages:
S. cerevisiae:
|
|
cyclin |
Cdk |
|
|
|
G1>>S phase |
Cln 1,2,3 |
Cdc 28 |
|
|
|
S-phase |
Clb 5, 6 |
Cdc 28 |
|
|
|
Replication origin firing |
Dbf4 Clb 5 |
Cdc 7 Ccdc28 |
Firing replication origin |
|
|
M-phase entry |
Clb 3,4 |
Cdc28 |
|
|
|
M-phase progression |
Clb1,2 |
Cdc28 |
|
|
|
M-phase exit |
Clb destruction |
|
|
|
|
|
|
|
|
|
Human and other Vertebrates:
|
Cyclins |
Cdk (protein kinase) |
Cyclin level |
Note |
|
Cyc-D1, D3 |
Cdk-4, 6 |
Increase |
START- G1 phase progression |
|
Cyc-E |
Cdk-2 |
E-Increase, D-decrease |
Onset of S phase, G1 >S |
|
Cyc-A |
Cdk-2 |
A-increase, E-decrease |
S-phase progression |
|
Cyc-A |
Cdc-2 (cdk-1) |
A-decrease, |
S through G2 |
|
Cyc-B |
Cdc2 (cdk-1) |
B-increase |
M-phase progression |
|
Cyc 13(Mr 45-47KD) |
Cdc2 (Mr 34KD) |
|
Prevents S-phase before M-phase |
|
Cig 2 |
Cdc2 |
|
Prevent the start of M-phase before S-phase completion |
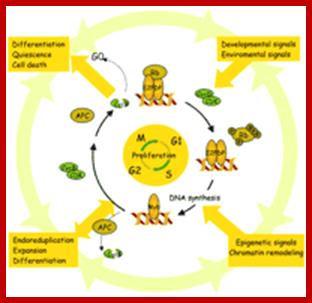
1. Regulation of Cell Proliferation is an Essential Process in the Establishment of Plant Architecture. The major checkpoints of the cell cycle are the G1/S and G2/M transitions. CDK/cycD complexes act at the G1/S checkpoint by phosphorylating the RB protein, causing the release of the E2F transcription factor and entry into S-phase. In the G2/M transition, active CDK/cycB complexes induce the entry into mitosis. Their APC-mediated degradation completes mitosis. The cell cycle machinery responds to external signals such as hormones, sucrose, and light, which are integrated with developmental, positional, and epigenetic signals. As a consequence, cells modulate their activity to maintain proliferation competence, become quiescent, expand, differentiate, endo-reduplicate, or die. The arrows represent the complex interconnections not only between the core of the cell cycle machinery and the different stimuli but also between expansion and differentiation or expansion and development. http://www.plantcell.org/
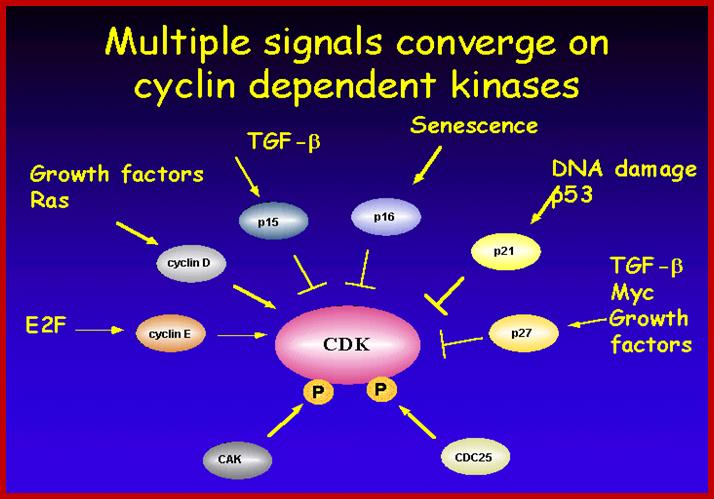
Multiple signala converge on CDKs some phosphorylate and some block phosphorylation. CDK can aso be phosphorylated by CAK and cdc25. Only growth factors have positive effects but many factors like TGF-b, p16, DNA damage, p21, p27 have negative effects. The figure presents a vivid picture of different signal has an effect or an affect on the activity of CDKs; CAK and CDC25-phospatae; www. streaming.cineca.it
E2F family consists of E2F-1 to E2F-5, they have great affinity to members of pRB, 107 and 130. E2F is the key downstream target of RB; binding of the E2F to a DP family member is essential for high affinity binding to E2F consensus sites and to RB family members.
It is important to note that the effector i.e., the protein kinase subunit, often called Cdc-28 in S. cerevisiae, or cdc2 in S. pombe, and CDK in other systems, is more or less same at all stages of cell cycle, but the cyclin, the partner varies from stage to stage, such as G1-cyclins, S-cyclins, G2-cyclins, M-cyclins and so on. When such cyclins combine with the cyclin dependent protein kinase, when active, for it becomes active, when the kinase subunit is phosphorylated at Thr.160 (or Thr 161 in other systems), they act on different targets at different stages of the cell cycle. Most of the cyclins are synthesized in temporal fashion, starting at the beginning of G1 and build up to M-phase and then they are degraded (by proteosome in ubiquitination mode), again in temporal fashion; and the timing of degradation is critical; so, cyclins act as regulators of the protein kinase, where kinase subunit is same and the cyclins are different. Thus, the regulation of cell cycle is regulated by the synthesis and timely destruction of the said cyclins.
Accessory factors:
Though cyclins and Cdks are considered as the prime factors in controlling cell cycle events, there are other factors, which are as important as cyclin-CDKs. There are several kinases (some are cyclin dependent and some are cyclin independent) and several phosphatases. They are-
Nim 1(Never In Mitosis): is a signal mediated protein kinase Inhibits wee1 by phosphorylation. Nim-1 pathaway pathway links to cdc2/cyclin system to external signals.
Wee 1: Is a kinase; inactivated by nim-1 by phosphorylation, Dephosphorylation makes it active’ when active it phosphorylates Cdk’s threonine 14 9 (or Tyrosine 15) and makes it inactive. Wee-1 activity is determined by signal input and signal transduction across the membrane.
CDK kinase (CAK): Phosphorylates Cdk’s active site threonine 160 (or Threonine 161).
Cdc25: It is a phosphotase,( counter part of this in the fly is “string” gene). Its Mr. is 80KD. It is active when phosphorylated and inactive when dephosphorylated. When Thr 14 (or Tyr 15) is phosphorylated the Cdk is inactive, but Cdc 25 dephosphorylates these sites; when this Dephosphorylation is coupled with the phosphorylation of Thr160 (Thr161) by CAK, Cdk-cyclin becomes active. The level of cdc25 reaches a threshold at M-phase, perhaps marks the end of S-phase.
CDK Inhibitors (CkIs):
Though cyclin-Cdks play critical regulatory roles in cell cycle, there is another set of molecules that regulate the regulators; in yeasts they are Cdk inhibitors or generally they are cell cycle kinase inhibitors (CKIs). There are different types of CKIs, such as far1p, Sic1p. The inhibitor binds to Cyclin-Cdk complex and prevents their activity. In metazoans, such molecules are called inhibitors of Kinase or Ink family of inhibitors. They bind to Cdk and exclude cyclin binding; they are called kinase inhibitors called ‘Kips’ and those that bind to cyclins and inhibit kinase activity are called ‘Cips’ (cyclin inhibitors).
MP kinase inhibitors (in the form of dimers) bind to kinases to form inactive complexes. Thus, they prevent phosphorylation Retinoblastoma proteins. So the cell cycle is checked at G1 or Go stage. CKIs classified into two classes-Inks and Kips.
CDK Inhibitors: There are CDK inhibitors such as INK family; p15, p16,p18,p19, They are specific ally associated with CDK4 and CDK6.
P21 family p21,27, 57 can inhibit all G1 Cyclin/CDK complexes and to lesser extent cyclinB/CDC2
INKs (Inhibitor of Kinase):
They are Cdk inhibitor proteins: INK 4 family is specific to Cdk4 and Cdk 6. Ink4 has four members- p15 (INK-4B), p16 (INK 4A), p18 (INK4C), and p19 (INK4D). They contain ankyrin repeat sequences. P16 and P19 bind next to ATP binding site, so prevents its catalytic activity. It also induces conformational changes so cyclin cannot bind. They act on cyclin D complexed either to Cdk4 or Cdk6.
Another class of inhibitors such as Sic I binds to Cdc28-clb2, in S. cerevisiae, inactivates the kinase at G1. So, entry of cell cycle into S-phase requires the degradation of Sic-I by ubiquitination mode. SCF acts as E3 ligase system. The Skp1-Cullin factor (SCF complex) consists of cdc53, Cdc4, Skp1 and Cdc34. These are involved in G1 cyclin destruction.
Kips: This is another class of inhibiters consists of P21, p27 and p57, they are identified by their molecular weights. They in general act on G1/S class Cdks. P21 binds to all Cdks- Cdk2, 4 and 6, thus block progress through all stages of G1/S. Increase in p21 concentration is inhibitory. Many a times in cultured cells one finds PCNA is also complexed with CDK-cyclin along with p21. so it controls G1/S stage progression. P27 also binds to Cdk-cyclin and blocks progression into S-phase, but it’s over expression leads the cell to go into Go stage.
Cyclin H/cdk-7: it is associated with TF-II H and involved in phosphorylation of CTD tail of RNA polymerase II; TF-II B also contains cyclin like helix bundles.
Cdc7-cyc-DBf4 kinase: It is serine/Thr protein kinase required for the onset of S-phase. The cyclin Dbf4 is constitutively synthesized but rapidly degraded from late M to G1. Activity peaks at the onset of DNA replication. Human homolog is Hsk (Homolog of Cdk Seven Kinase, however CDK lacks PSTAIRE sequence. The target of this complex is Mcm2. Loading of Mcm2 on to ORE region is important in triggering the firing replication origin.
Cdk Activating Kinase (CAK): Cdk 7-cyclin H has CAK activity. Cyclin-A binding to cdc2 (homolog iscdc28) exposes active site and ATP binding site in Cdk protein, where Thr 160 (Thr 161) is made available for CAK to act upon. CAK phosphorylates Thr 160 (161) of Cdk to make Cdk-cyclin A to be active.
Positive regulation of Cdk by cyclins is often counterbalanced by negative regulation by Inks, Cips and Kips.
Rum 1 protein: Cdc2/cdc13 MPkinase is influenced by Rum-1 factor. When rum-1 is over expressed cell does not enter M-phase, but s-phase goes through multiple cycles. When rum1 is deleted the cell enters M-phase prematurely. This is expressed between G1 and G2 and keeps the MPK inactive. So this is essential for the S-phase to proceed.
Nucleophosmin: It is a protein, in unphosphorylated form binds to centrosome at the end of M-phase and prevents duplication of centrosome. But Cdk2/cyclin E phosphorylates Nucleophosphomin, at M-phase. The phosphorylated Nucleophosphomin then dissociates from centrosome. This is further augmented by Calcium mediated calmodulin dependent kinase II activity at G1-S boundary facilitates the duplication of centrosome. Centrosome duplication is essential for the organization mitotic apparatus.
APC complex:
It is called Anaphase Promoting Complex: This is multisubunit complex made up of eight proteins; the complex is also called Cyclosome. Such complexes are found in yeast and animal tissues. APC becomes active during M-phase. It functions as E3-ligase in ubiquitinated proteosome mediated protein degradation. First the proteins Cdc20 and then Cdh-1 displaces cdc20 and binds to APC and activate its ubiquitination activity. Cdc20-APC is essential for the degradation of Securin, which paves the way from Metaphase to Anaphase
· Once activated, MPKs initiate M-phase, the progress of it takes its own course and it does not require active MP kinase anymore, so to exit from M-phase MP kinase has to be inactivated. One way to inactivate is to block the catalytic site by inhibitors, or dissociate Cyclin from the kinase, or phosphorylate Thr14 (or Tyr15) or destroy cyclin the CDK partner. Actually, as the M-phase sets in the first cyclin to be destroyed is cyclin-A at Metaphase. Then little later i.e. at Anaphase, Cyclin-B is degraded by ubiquitination mode, making MP kinase inactive. This type of degradation mediated by Cdh1 activated APC complex (Cdh1 is essential for the degradation of Clb 2 which are B-like cyclins); this paves the way for the cell to exit from Mitosis.
When chromosomal DNA replicates, single stranded chromosome becomes double stranded, for reasons of stability, a protein complex called Cohesins glue the two strands to each other. But when they reach equatorial region or little earlier, the tightly held chromosomal strands release from one another, yet they are still held at centromeric region. For equal segregation of chromosomes, the kinetochore complex has to split and free chromosomal strands from one another, so the strands can move to their respective poles. For the chromosomal strands to free from one another, glue called Cohesin complex that holds chromosomal strands, is degraded, so at Anaphase chromatin strands separate. In some systems chromosomal strands are freed at the end of prophase itself, but centromere is still held together, in such cases kinetochore complex splits by the protease activity induced by APC.
· The APC complex that targets cohesin complex and kinetochore complex is activated by cdc20. It is activated at M-phase or little earlier and performs destruction of Securin (pds-1p), which triggers the release of two chromosomal strands from one another. This process is critical for the separation of chromosomes at anaphase, so the complex is called Anaphase Promoting Complex.
Cohesins and Condensins:
Cohesins and condensins are heteromeric proteins made up of smc proteins (Structural Maintenance of Chromosomes) and non-Smc proteins. Cohesins are made up of two smc proteins, Smc-1p and Smc-3p and two non-Smc proteins, Sec1p and Sec3p; where as condensins are made up of Smc2p and Smc4p and Sec2p and Sec4p. Cohesins are responsible for holding two sister strands together as parallel strands all along the length including kinetochore region. But condensins are responsible for the condensation of chromatin from long convoluted threads into short and stable threads at metaphase.
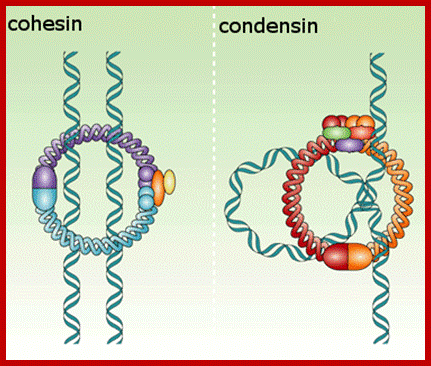
Models ofCondensins and Cohhesins; www.nature.com
Cohesins: They are complex of proteins, made up of Smc1 & 3 and Scc1P & Scc3p. Smc1 and Smc3 of cohesins are coiled coils with a flexible hinge. In Smc1 and Smc3 coiled coil protein pairs show V-shaped bending with 86 ^o apart. But the Smc2 and Smc 4 proteins when dimerizes the flexible angle is steep of 8^o.
When Smc1 and Smc3 dimerize parallel to each other they are oriented in antipolar fashion. At either ends they have a DNA binding domain and a domain for the binding of ATP. One end of the coiled coils bind to the DNA and the other end of the protein dimerizes with another Smc protein pair. Two such Smc pairs can hold on to the same DNA at one site and at the other end is free for dimerization with another Smc protein pairs that is anchored on to another DNA. If two sets of Smc protein pairs, one holding one strand and another holding the opposite strand. When the free ends are paired and linked by Scc1p and Scc-3p, two chromosomal strands will be held parallel to each other. Several protein pairs all along the length of chromosomes provide such links, thus chromosomal strands are paired and glued. Here the glue is Scc1p and Scc3p, perhaps one more protein pds5 may also be present. Degradation of this makes chromosomal strands to separate from one another.
Condensins: They are also made up of a complex of proteins, such as Smc 2 and Smc4 and two non-Smc proteins called Scc3 and Scc4. Here the Smc proteins 2 & 4 coil to each other in antiparallel fashion. The flexible portion can generate an 8^o angle. These proteins bind to the same chromosomal DNA at two sites i.e., one pair at one site and the other pair at another site, perhaps at a distance. The free ends can dimerize. When Scc2 and Scc4 proteins dimerize the other ends, which are bound to chromosomal DNA; the DNA found in between is looped out. Many such condensins all along the length of chromosomes act on at different sites and condense chromosome to a maximum at metaphase. Whether or not there is any relation between Histone-1 phosphorylation induced condensation and Condensin operated condensation, is not clear; sure, there should be a relation
Operation of Cell Cycle:
Cells, irrespective of their ploidy divide either during growth or during reproduction. During reproduction, a diploid gamete-producing cell undergoes reduction or meiotic division. But a similar diploid or haploid cell during growth and development goes through a series of mitotic cell divisions. Even a fully grown organism, where most of the cells at all times are in resting or what is called Go state, occasionally undergo cell division in order to compensate cell loss. In culture condition cells also undergo cell division to multiply in numbers.
· In general cells in an environment provided with rich nutrients divide and redivide e.g. yeast, but cells under culture conditions initiate cell division when they are stimulated by mitogens. Cells in a tissue require stimulation for division. At that time cell size increases and when the cell mass reaches an optimum level to its volume, it initiates cell division, if it is somatic it is called Mitosis.
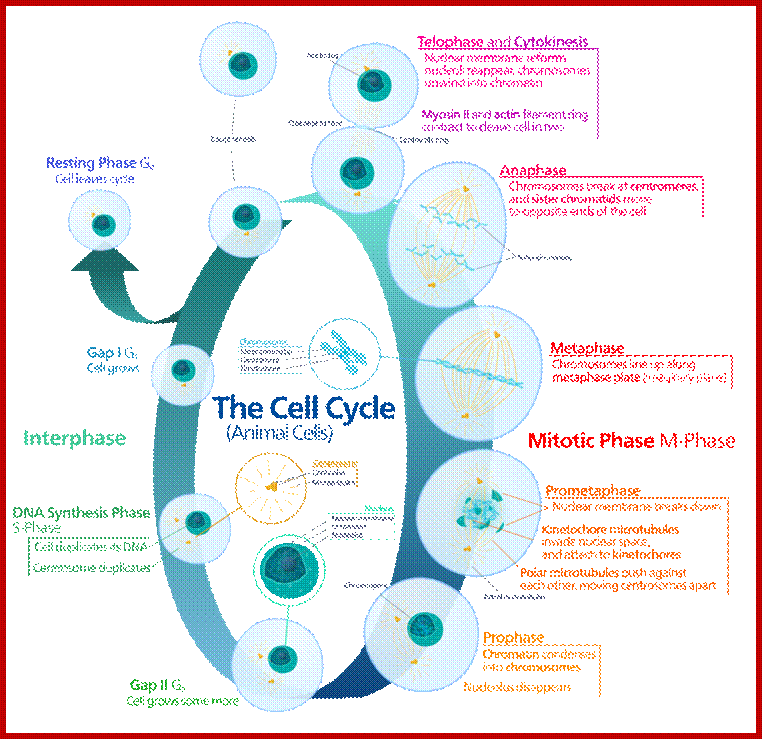
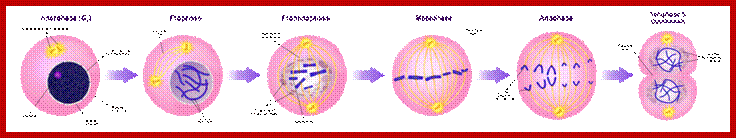
Mitosis goes through several physical and biochemical changes in the form of stages or phases. Mitosis has several stages such as M-phase and Interphase. Between two M-phases there exist an intervening phase called Interphase. The M-phase its self consists of sequential steps like Prophase, Metaphase, Anaphase and Telophase and finally Cytokinesis culminates in producing two daughter cells, which have inherited their genetic material equally. Interphase, in general occupies longest time in cell division. It can extend to 10-12 hrs in a 24 hr cell cycle. But the M-phase takes just 30 minutes or 1 hr.
· Interphase when in resting phase exists in what is called Go stage, where all cell cycle processes are shut down. But when the cell is stimulated, either by nutrient supply or by mitogens, they renter from Go stage and enter into G1 stage. The G1 is a preparatory stage for the next phase called S-stage. In the S-stage the chromosomal DNA replicates and generates two copies of them. Then the cell enters into another stage called G2 stage; which is again another preparatory stage for M-phase. G1, G2 are called so scientists did not know what exactly happen at these intervening stages, so they called it G1 and G2, which is a Gap in the knowledge about them. Though these stages are sequential and temporal, they don’t enter to the next stage until and unless each of the stages has completed their requirements and functions. To prevent any such precautious entry into the next stage, they use checkpoints, which act as control loops where cellular events should be completed at the earlier stage to move to the next stage, otherwise they remain in the same stage till all the events required are completed.
G1 Stage:
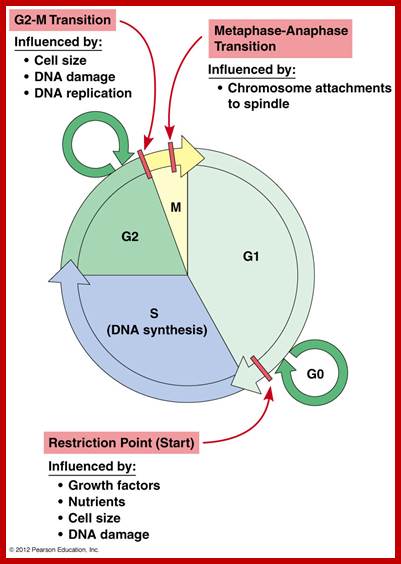
The G1 occupies approximately 10 to 12 hr where the cell prepares for S-phase. Important check point here is called START point or restriction point, once it passes through there is no going back.

E2F is a transcription factor that activate transcription required for DNA replication, but it is blocked by RB; then specific Cyclin-CDK phosphorylate RB; this releases E2F leading to DNA replication, www.journals.cambridge.org
Once cells are stimulated, they measure cell mass and cell volume. There are genes, which do this function. Once cell mass to cell volume is measured and full filled, it launches into a series of molecular events that sets the stage to next stage. In general, at G1 stage, inputs for DNA replication are shut off. This is achieved by sequestering all those required Transcriptional Factors (TF-IIs) required for activating genes that are essential as inputs for initiating and executing DNA replication to completion. The factor that blocks is RB protein, which was identified as a mutant gene causing Retinoblastoma disease (a cancer). In its native state RBs sequester all those transcriptional factors -E2Fs. These are required for activating genes for cyclins. Once cyclins are synthesized, they activate certain cyclin dependent kinases (Cdks). These are blocked by RB proteins.
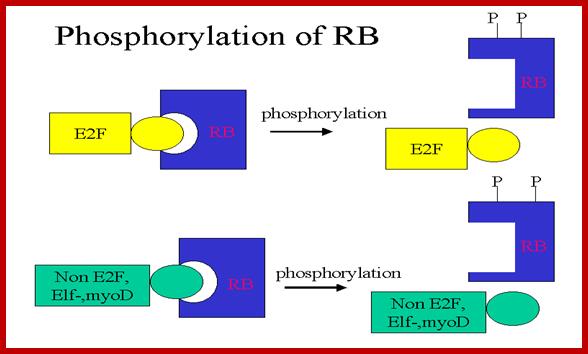
RB binds to transcription factors like E2Fs and some non-E2Fs, which are sequestered when RBs are non-phosphorylated, but when, phosphorylated they are released for activating S-phase specific gene expression.www.images.1233.tw
RBs are phosphorylated at specific sites. Phosphorylation of RBs by different combination of Cdc-Cdks at different stages; by binding to RB they inactivate RB protein.
· Earlier signal transducing cellular events triggered by mitogens
or nutritional factors do cause specific phosphorylation and dephosphorylation reactions.
All most all cells have a battery of thousand or more Kinases and equal number or more phosphatases, which act specifically on their targets; thus they activate or inactivate certain substrates, which can be a protein or a carbohydrate or any other target cellular component.
When the cell is still in earlier G1 stage the Cdks are phosphorylated at Thr 14 (tyr15) and rendered inactive by wee1 protein kinase (wee1 is active when it is dephosphorylated and inactive when it is phosphorylated; this is controlled by nim1 ‘never in mitosis’ protein). The Cdk has another site for phosphorylation, i.e. is Thr 160 (Thr161), which is located next to Kinase active site, and ATP binding site at the C-terminal part of the Cdk, which actually folds back over kinase active site when its Thr 160 is not phosphorylated. If this Thr 160 is not phosphorylated cyclin cannot bind and make Cdk-cyclin complex active. At earlier G1 stage the Cdk is rendered inactive. As the cyclins build up, another protein level increases; it is cdc25 and it is a phosphatase specific to Cdk Thr 14 (Tyr15). The buildup of cyclin synthesis starts at early part of G1 stage and continues to build till M-stage, at which time it is degraded abruptly. Cyclin protein have rapid turnover, their half-life is just 15 minutes or so.
Thus, RBs and similar proteins bind to E2F factors, thus transcription of genes required for DNA replication are kept in inactive state. Another event that keeps Cdk inactive is by phosphorylation of Thr14 (Tyr15) by Wee1.
· After cell stimulation, cyclins (mostly cyclin-D) build up, Cdc25 also builds up. At this stage Cdc25 dephosphorylates Cdks Thr-P14 (Tyr15-P). At the same time another protein kinase called Cdk activating enzyme called CAK phosphorylates Thr160 (Thr161). Almost at the same time Cdk inhibitor proteins that are bound to Cdk-cyclin complex are also released. These biochemical events facilitate the binding of G1 cyclins (cyclin-D) to Cdk properly and the complex becomes fully active.
As the Cdk-cyclin complex in its fully active state phosphorylate RBs and its associated proteins, thus make E2F factors released free. These transcription factors with their associated DPs (Dimer proteins) bind to their respective promoter elements and activate the transcription of genes, whose products required for more cyclin synthesis and factor and components for DNA replication. Once the said factors synthesized in sufficient amounts cell enters into S-stage. At this stage several cyclins are synthesized, such as cyclin E and others for M-phase activity
S-Stage: S-phase is of short duration of 6-8 hrs. This is most precise and exact process and its execution should be error free. S-phase initiation is again contr0olled by another set of factors. Until and unless they are made available DNA replication is not initiated.
For the replication of DNA, replication origins have to be fired, that is they have to open into replication bubbles. In eukaryotes DNA is compacted by nucleosomal organization into higher order of compaction i.e. chromosome. Chromosomes at this stage have to be relaxed and origins should be made available.
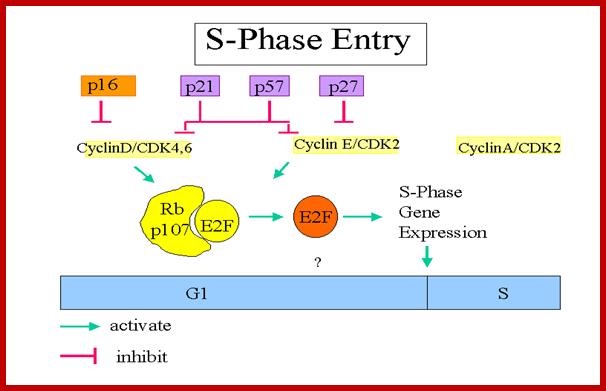
P16, p21, p57, and p27 are cyclin-CDK blockers; Cyclin D/CDK4,6 phosphorylate thus RB bound E@F gets released, then E2F activate genes required for DNA replication; Entry into S-phase is critical, but it is governed by and regulated by the Cdc-Cdk inhibitors and RBs. The release of E2Fs and other non E2F TFs is critical for the entry of the cell into S-Phase; teach.med.ncku.edu.tw.
· Eukaryotic DNA is long (~3.2x10^9 bp or so), and linear, unlike E. coli, which is circular. The thousands of origins are located in what is called replication initiator zones, ithin in which the initiator zone contains several origins. On the basis of yeast’ ARS sites, most of the origins contain a 11 bp long ORE (Origin Recognition Elements) with specific sequences. Next to it there are DNA unwinding elements called DUE. On either side of these two elements there can be auxillry sequences.
At the time of initiation of replication, the ORE should bound by a complex of proteins called ORC- origin recognition complex (> 400KD). For firing the replication origin it requires Mcms (mini chromosome maintenance proteins; they are hexamers; they are acquired when the nuclear membrane is dissolved. This happens only once in one cell cycle at M-phase. Cdc6 proteins are also acquired at this phase; together they act as licensing factors. At the same time, two more factors are acquired; they are Cdt1 and Geminin. The Geminin prevents the loading Mcms second time before completing M-phase. Loading of Mcms is crucial for it actually opens the origin region into replication bubble. Cdc6 performs this only when it is phosphorylated, which is performed by Cdk-S1 cyclins? Once replication is initiated Mcm and Cdc6 are released from the origin, but CDC complex remains bound to the origin.
· When all the inputs are made available DNA replication progresses to completion; then only the cell enters G 2 stage. If during replication any error or damage is occurs, cell cycle halts and starts repairing the damage. Even the cell enters to G2 it still waits till it is repaired. If the damage is beyond repair, p53 protein, which act as a sensor for DNA damage activates the genes for the synthesis of P21 and such proteins, which bind to Cdk-cyclin complexes and halt the cell cycle progress. Progress of S-phase is greatly facilitated by s-phase cyclins or cyclin-E, but once the S-phase is initiated s-cyclins get degraded. The next cyclin expressed is Cyclin-A from S to G2 stage.
G2 Stage: Again it is phase waits for all the inputs required for M-phase. If the DNA is damaged it waits in G2 stage till the damage is repaired, otherwise the cell is signaled to suicidal death or Apoptosis.
M-phase: this stage shows lot of physical changes like disassembly of nuclear membrane little early to Metaphase and pore complexes, duplication of centrosome, appearance of mitotic apparatus emanating from MOTC at poles, chromosomal strand separation from one another that are bound by Cohesins, this includes splitting of kinetochore, chromosomal condensation, attachment of tractile fibers on to kinetochore complexes, movement of sister strands to opposite poles headed by kinetochore tractile fibers, , dissolution of all membrane components into vesicles, depolymerization of microtubules into tubulins, reorientation of actin filaments, destruction of many proteins including cyclin-A and cyclin-B.
· M-phase is activated by Cdk-cyclin-B complex. During G1 and S-phase cyclin accumulates in cytoplasm, when it reaches a threshold, gets activated and moves in to the nucleus, where Cdk subunit undergoes dephosphorylation at Thr14 (Tyr15) by cdc25 phosphotase, phosphorylation of Thr160 (Thr 161) by CDK activating Kinase CAK. Now cyclin-B binds to CyclinA-CDK2; this complex is often called MPF, or MPK. This activated CDK2-cyclin-A complex targets many proteins for phosphorylation such as chromosomal H1, nuclear Lamins, centrosome, microtubules and yet many unknown structures.
It is at this stage the MP-kinase activates Anaphase promoting complex (APC). Activated APC combines with specific adaptors such as cdc20 and targets Cohesin’s complex, where it targets Securin and degrades Securin. The Securin always keeps Separin sequestered. Separin is an endopeptidase or call it endo-protease. Degradation of Securin releases Separin, which now acts of Scc1 and Scc3 which are the components of Cohesin protein complex, thus the glue is dissolved and chromatin separate from one another, also the kinetochore by some unknown sensing mechanism also splits and facilitates the anaphase movement of chromosomal strands. APV complex as described earlier, is ligase system targets proteins for ubiquitinated proteosome mediated digestion
· The APC system gets replaced with another adaptor protein called Cdh1; this then targets; first cyclin A and then Cyclin-B. Mitotic Cdk-cyclin complexes prevent reinitiation of S-phase DNA replication and also prevent second round of Mitosis before DNA replication.
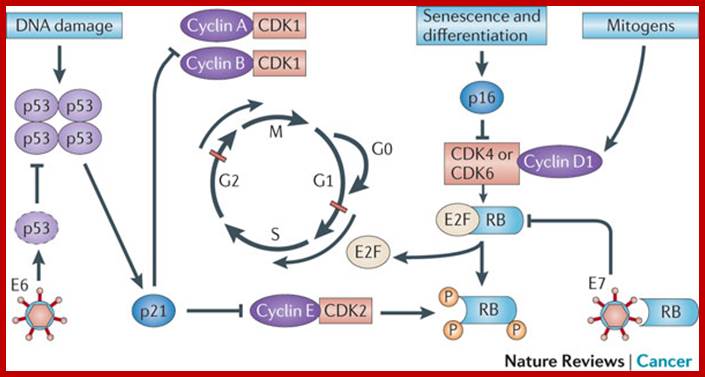
Cell cycle deregulation; The molecular biology of head and neck cancer;
The cell cycle is regulated by complexes of cyclins and cyclin-dependent kinases (CDKs), some of which are indicated. In addition, there are various important inhibitors of these cyclin–CDK complexes. To allow cell cycle progression, cells have to pass the G1 restriction point (red bar) that is controlled by the retinoblastoma pocket proteins, RB, p107 (also known as RBL1) and p130 (also known as RBL2). Only RB is indicated, but the other pocket proteins have similar activities. These normally bind to and inactivate the E2F transcription factors, which induce the expression of S phase genes. In response to a mitogenic signal, the cyclin D1–CDK4 and cyclin D1–CDK6 complexes are activated. These phosphorylate the Rb pocket proteins, causing release (and therefore activation) of E2Fs. Induction of cyclin E by E2F and subsequent additional phosphorylation of RB by the cyclin E–CDK2 complex initiates entry into S phase. The inhibitor for the cyclin D1–CDK4 and cyclin D1–CDK6 complexes is p16INK4A, which is encoded by CDKN2A, a gene in theINK4A locus at chromosome 9p21. The expression of p16INK4A mediates senescence and differentiation. The interplay between the cyclins, CDKs and their inhibitors determines whether the restriction point can be passed, and a growth factor stimulus is usually required. A second important control mechanism of the cell cycle occurs during G2 phase, when the DNA has been replicated and replication errors are repaired. The key protein involved in the response to replication errors and other DNA damage is p53, which is usually maintained at low concentrations by MDM2-mediated degradation (not shown). DNA-damage sensors, including ataxia-telangiectasia (ATM) and ataxia-telangiectasia and Rad3-related (ATR), phosphorylate the checkpoint kinases CHK1 and CHK2, leading to increased p53 activity by phosphorylation of various downstream molecules, including p53 itself (not shown). The p53 tetramers act as a stress-induced transcription factor and induce the expression of p21CIP (also known as CDKN1A), which inhibits several cyclin–CDK complexes and halts the cell cycle. Besides its crucial role in cell cycle control, p53 is also a master regulator of apoptosis and many other stress-associated cellular functions, and is therefore one of the main targets for inactivation in many cancers. The human papillomavirus (HPV) genome contains various early and late open reading frames and encodes two viral oncoproteins: E6 and E7. The E6 protein binds p53 and targets the protein for degradation, whereas the E7 protein binds and inactivates the Rb pocket proteins. The molecular consequence of the expression of these viral oncoproteins is cell cycle entry and inhibition of p53-mediated apoptosis, which allows the virus to replicate. In a 'productive infection' the expression of E6 and E7 is confined to the differentiating layers of the squamous epithelium of the cervix and virions are produced. An oncogenic infection is associated with E6 and E7 expression in the basal layer (where the stem cells reside) and causes abrogation of the cell cycle checkpoints. C. René Leemans, Boudewijn J. M. Braakhuis & Ruud H. Brakenhoff http://www.nature.com/
Regulation of Cell Cycle:
As in prokaryotes, Eukaryotic DNA replication is restricted to either Mitosis or Meiosis stages. Mitosis is used for growth and development and in some lower forms it is one of the modes of reproduction. But meiosis is mostly involved in reproductive stages. Whether Mitosis or Meiosis, cell division is highly regulated and precise and exact. Mitosis goes through several stages such as Prophase, Metaphase, Anaphase, Telophase and cytokinesis (not always) and then enters Interphase, which is an intervening stage at which the cell prepares for the next division or goes into resting phase where the cells undergo differentiation to specific cell type.
The above photomicrograph shows yeast S. pombe is going through cell division.
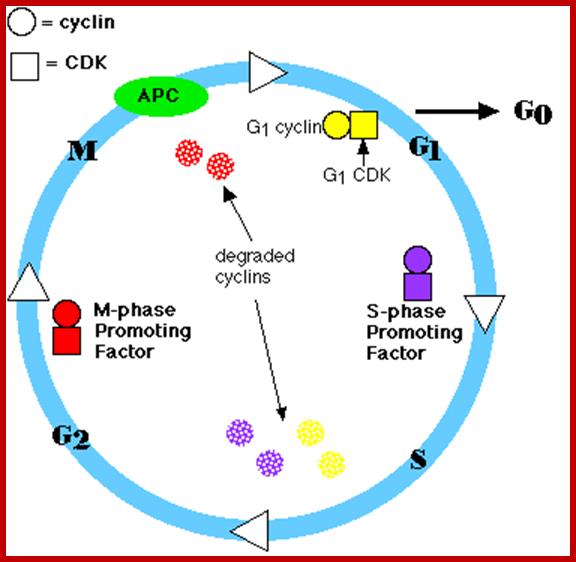
Overal role of Cyclin-CDK proteins; www.biology.kenyon.edu
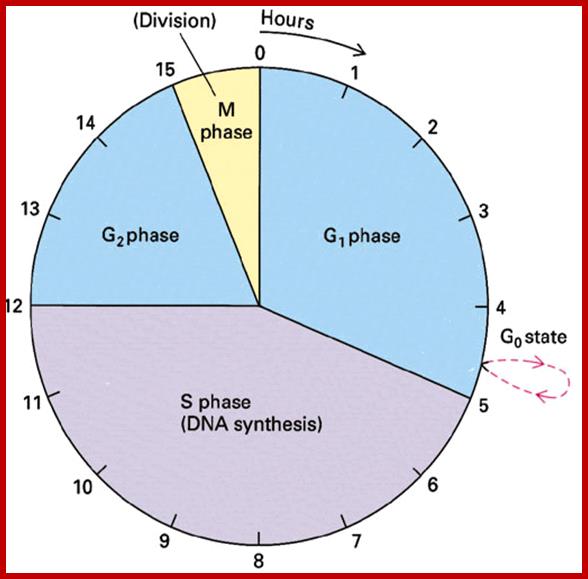
The diagram shows the time required by each of the phases.
www.uic.edu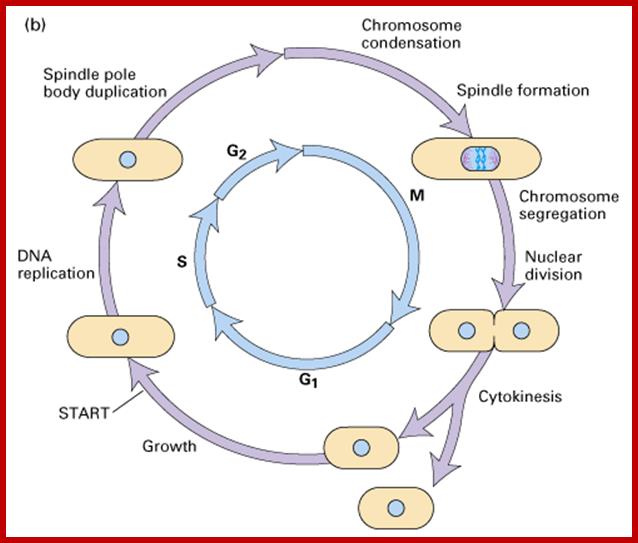
- Among the five stages in a 24hrs cell cycle animal cell culture, Interphase occupies longest period where as the other stage called M-stage lasts just about 0.30 min to 1 hr. But in embryonic stages the cell cycle is just 15 –30 minutes or less; in this the cell after M-phase, enters directly to S-phase. It also means all the inputs required for the next stage already exist in embryonic cells. In S. cerevisiae, G1 last for 15 min, S-phase requires 30 min and M-takes about 75 min all together require 120 min. While S. pombe (fission yeast), G-1 requires 20 min, S takes 20 min, G2-takes 35 min and M-phase lasts for 45 min, I n all 120 min. In both the above-mentioned forms cell divides without dissolution of nuclear membrane.
Interphase consists of sub-stages such as G1, S and G2; where, G at earlier times stands for gap in the knowledge about these stages. In 24 hr cell cycle events G1 occupies 10-12 hrs, S-stage about 6-8 hr and G2 stage 4-4.5 hr. The G1 phase is considered as preparatory phase for DNA replication, but the Cells escape from G1 phase in terminally differentiating cells into what is called Go stage, where cells assume specific shape, structure and function. But some of the cells remain embryonic and such cells can be stimulated to become dividing cells by some mitogens, and they can be differentiated depending upon the kind of stimulus they get, or stimulus provided. They are called STEM cells. Such cells are found not only in animal tissues but also in plant tissues, in fact plant cells have greater potentiality to be mitotic. Such cells can be stimulated by mitogens to enter into cell division mode, where they enter again into G1 phase. The S-phase is for DNA replication and G2 stage is a preparatory phase for M-phase, where the nucleus disassembles, chromatids separate and mitotic apparatus assembles, the centromere split, sister chromatids are pulled to their respective poles, daughter nuclei reform and cytokinesis leads to division of cytoplasm into two cells. This is a simplistic description of cell division.
- Each of these phases are regulated by specific molecular events and factors, until each of the events in each of the phases are completed cell won’t enter in to next stage. Thus, the entry and exit to and out of stages or phases is tightly controlled by a variety of factors; and the factors themselves undergo changes in their turn over (synthesis and degradation), activation and inactivation of them.
The major checkpoints lie in between G1 and S phase and G2 and M-phase and another control point exists within the M-phase events at anaphase. Notwithstanding the said checkpoints, DNA damage can introduce a checkpoint, where until the damage is repaired, cell does not enter M-phase, this can happen at S-phase or at G2 phase; if the damage is beyond repair the cell is signaled for Apoptosis.
Checkpoints act at transition points where all the earlier events have to be completed before it progresses to the next stage. They also act as surveillance systems. It is regulatory loop where initiation of one event depends on the completion of the earlier event, so progression through a checkpoint is strictly controlled.
- Among the many cellular components involved cyclin dependent kinases (Cdks) play a significant role. Cells employ more than 1000 kinases and also employ equal number of phosphatases. The beauty of the interplay of these two components is that many proteins and other cellular components rendered active when they are phosphorylated at specific sites on them. Some of them get inactivated when they are phosphorylated, but some, depending upon the individual component, become active when they are dephosphorylated. Thus, Specific kinases phosphorylate specific proteins or similar substrates at specific sites; thereby they activate or inactivate cellular components. Phosphatases in turn remove phosphate groups from specific sites in specific protein, so the substrate may be rendered active or inactive. Similarly, there are a host of inhibitors especially kinase inhibitors, and they play a pivotal role.
- In all this, the entry of cells into S phase is very crucial for the cell to divide and generate two individual cells with the genetic material equally duplicated and equally distributed without making not even a single mistake, is very very important.
- Thus DNA replication at S phase is critical. For initiating DNA replication exactly at S-phase and completion of it in S phase requires an input of many qualitatively different factors that have to be provided just before it. So DNA entry into replication mode, completion of replication and separation replicated daughter DNA molecules in all its glory is controlled by the inputs of several factors. The diagram below shows some of the crucial components required and events that rigger the initiation of Replication at specific sites and at specific time is depicted.
- The S-phase depends on M-phase; till is over another round of DNA replication won’t be initiated
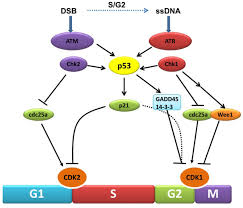
Chekpoint regulation by the DDR: ATM and ATR orchestrate a transitory delay of the cell cycle in response to DSBs and ssDNA, respectively. Whereas, direct phosphorylation of of cdc25a and wee1 allow a rapid establishment of the G1/S and G2/M checkpoints, p53-dependent regulation contributes to checkpoint maintenance at later timepoints. During S and G2 phases DSBs can be resected leading to the generation of ssDNA, which also activates ATR-signaling. The diagram is an elaborate depiction of various components assembling and disassembling at specific stages of G1 and S-phase. The critical components are ORC, MCMCdc6 and Cdt1. Components like SCF, p21 and p53 have controlling power over the said events. www.intechopen.com
The G1 checkpoint, also known as the restriction point in mammalian cells and the start point in yeast, is the point at which the cell becomes committed to entering the cell cycle. As the cell progresses through G1, depending on internal and external conditions, it can delay G1, enter a quiescent state known as G0, or proceed past the restriction point. The decision to commit to a new round of cell division occurs when the cell is stimulated with mitogens and then the cell activates cyclin-CDK-dependent transcription which promotes entry into S phase.
During early G1, the transcriptional repressors Rb (retinoblastoma), p107 and p130, known as pocket proteins, bind to the E2F transcription factors to prevent G1-to-S transition. Rb binds and represses activator E2F transcription factors (E2F1-3), while p107 and p130 bind E2F4 and E2F5 to form complexes which repress transcription of G1-to-S promoting factors. Upon the decision to progress past the G1 checkpoint, cyclin D levels rises, and cyclin D forms a complex with CDK4 and CDK6, which in turn phosphorylate the pocket proteins. Phosphorylation of the pocket proteins causes the release of their bound targets, thereby relieving the repression of the E2F1-3 activators and translocating repressors E2F4 and E2F5 from the nucleus to the cytoplasm. This results in the transcriptional activation of downstream targets, which promote the G1-to-S transition, including another cyclin, known as cyclin E, which forms a complex with CDK2. The formation of the cyclin E-CDK2 complex then promotes a positive feedback loop which creates an “all or nothing” switch from which the cell can not return.[7] Following entry to S-phase and initiation of DNA replication, S-phase cyclin A, a transcriptional target of E2F1-3, forms a complex with CDK2 which phosphorylates E2F1-3 and prevents its ability to bind to DNA, thus forming a negative feedback loop. In another negative feedback loop, E2F1-3 promotes the transcription of E2F6-8, which in turn represses G1-S transition.
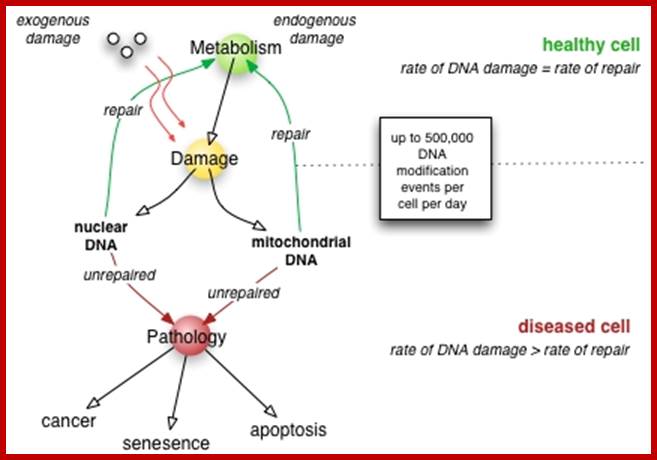
DNA damage chromatin structure transcription-coupled DNA repair single-strand DNA breaks DNA loop nucleosome ;http://blog.naver.com/; Nikolay A.Pestov et al; http://thejupital.com/
When DNA damage occurs, or when the cell detects any defects which necessitate it to delay or halt the cell cycle in G1, arrest occurs through several mechanisms. The accumulation of damage, to be specific, double-strand breaks or adducts stalling the replication forks, are among known stimulation signals for a global response to DNA damage.[ The rapid response involves phosphorylation events that initiate with either kinase ATM (Ataxia telangiectasia mutated) or ATR (Ataxia Telangiectasia and Rad3 related), which act as sensors, depending on the type of damage. These kinases phosphorylate and activate the effector kinases Chk2 and Chk1, respectively, which in turn phosphorylate the phosphatase Cdc25A, thus marking it for ubiquitination and degradation. The Cdc25A activates the previously mentioned cyclin E-CDK2 complex, by removing inhibitory phosphates from CDK2. In the absence of Cdc25A, cyclin E-CDK2 remains inactive, and the cell remains in G1. To maintain the arrest, another response is initiated, by which Chk2 or Chk1 phosphorylate p53, a tumor suppressor, and this stabilizes p53 by preventing it from binding Mdm2, a ubiquitin ligase which inhibits p53 by targeting it for degradation. The stable p53 then acts a transcriptional activator of several target genes, including p21, an inhibitor of the G1-to-S promoting complex cyclin E-CDK21. In addition, another mechanism by which p21 is activated is through the accumulation of p16 in response to DNA damage. p16 disrupts cyclin D-CDK4 complexes, thus causing the release of p21 from the complexes, which leads to the dephosphorylation and activation of Rb, which allows Rb to bind and inhibit E2F1-3, thus keeping the cell from transitioning to S phase.[8] Recently, some aspects of this model have been disputed.
The G1 stage, as said earlier, is the stage where cell prepares for DNA replication. The cyclins required at this stage are cyclins D. The required components for DNA replication, besides a large nucleotide and histone pools, are of DNA replication machinery, such as DNA polymerase, helicases, SSBs, and many other factors. It is during this stage or at the end of this stage transcription of genes required for DNA replication is activated. Transcription of the said genes requires transcription factors and their activation is sine quo non for the entry of the G1 to S-phase. If there is a damaged DNA at G1 stage entry into S-phase is prevented by the mediation of p53 and its associated components. If and only if all the required components for replication are provided then cell enters into S-phase.
Events: During early G1: The transcriptional repressors Rb (retinoblastoma), p107 and p130, known as pocket proteins, bind to the E2F transcription factors to prevent G1-to-S transition. Rb binds and represses activator E2F transcription factors (E2F1-3), while p107 and p130 bind E2F4 and E2F5 to form complexes which repress transcription of G1-to-S promoting factors. Upon the decision to progress past the G1 checkpoint, cyclin D levels rise, and cyclin D forms a complex with CDK4 and CDK6, which in turn phosphorylate the pocket proteins. Phosphorylation of the pocket proteins causes the release of their bound targets, thereby relieving the repression of the E2F1-3 activators and translocating repressors E2F4 and E2F5 from the nucleus to the cytoplasm. This results in the transcriptional activation of downstream targets, which promote the G1-to-S transition, including another cyclin, known as cyclin E, which forms a complex with CDK2. The formation of the cyclin E-CDK2 complex then promotes a positive feedback loop which creates an “all or nothing” switch from which the cell can not return.[7] Following entry to S-phase and initiation of DNA replication, S-phase cyclin A, a transcriptional target of E2F1-3, forms a complex with CDK2 which phosphorylates E2F1-3 and prevents its ability to bind to DNA, thus forming a negative feedback loop. In another negative feedback loop, E2F1-3 promotes the transcription of E2F6-8, which in turn repress G1-S transition.
When DNA damage occurs, or when the cell detects any defects which necessitate it to delay or halt the cell cycle in G1, arrest occurs through several mechanisms. The rapid response involves phosphorylation events that initiate with either kinase ATM (Ataxia telangiectasia mutated) or ATR (Ataxia Telangiectasia and Rad3 related), which act as sensors, depending on the type of damage. These kinases phosphorylate and activate the effector kinases Chk2 and Chk1, respectively, which in turn phosphorylate the phosphatase Cdc25A, thus marking it for ubiquitination and degradation.
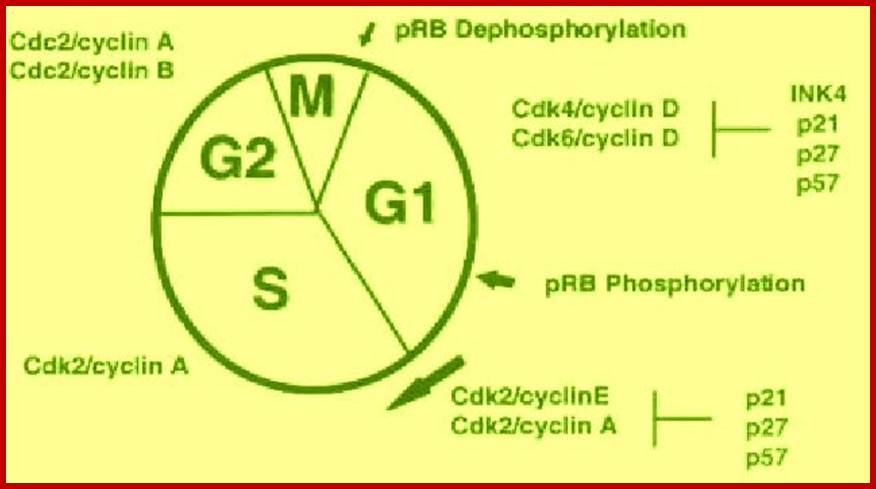
The diagram shows the kind of cyclin-Cdks involved in specific stages.

The retinoblastoma protein (Rb) is also a famous tumors suppressor protein. Rb binds to the activation domain of E2F and then actively represses the promoter by a mechanism that is poorly understood. It has been recently reported that Rb associates with a histone deacetylase, HDAC1, through the Rb 'pocket' domain. Rb cooperates with HDAC1 to repress the promoter of the gene related to cell-cycle. Active transcriptional repression by Rb may involve the modification of chromatin structure; http://www.nature.com/, www.cyclex.co.jp;
The Rb protein is a tumor suppressor, which plays a pivotal role in the negative control of the cell cycle and in tumor progression. It has been shown that Rb protein (pRb) is responsible for a major G1 checkpoint, blocking S-phase entry and cell growth. The retinoblastoma family includes three members, Rb/p105, p107 and Rb2/p130, collectively referred to as 'pocket proteins'. The pRb protein represses gene transcription, required for transition from G1 to S phase, by directly binding to the transactivation domain of E2F and by binding to the promoter of these genes as a complex with E2F. pRb represses transcription also by remodeling chromatin structure through interaction with proteins such as hBRM, BRG1, HDAC1 and SUV39H1, which are involved in nucleosome remodeling, histone acetylation/deacetylation and methylation, respectively. Loss of pRb functions may induce cell cycle deregulation and so lead to a malignant phenotype. Gene inactivation of pRB through chromosomal mutations is one of the principal reasons for retinoblastoma tumor development. Functional inactivation of pRb by viral oncoprotein binding is also shown in many neoplasias such as cervical cancer, mesothelioma and AIDS-related Burkitt's lymphoma.
The Rb gene is functionally inactivated in most human neoplasms either by direct mutation/deletion, such as in retinoblastoma, osteosarcoma and small-cell lung carcinoma, or indirectly through altered expression/activity of upstream regulators; responsible to check G1 phase b lock entr into S-phase. The Rb gene family includes three members, Rb/p105, p107 and Rb2/p130, collectively referred to as 'pocket proteins' over expression of them arrest cell at G1
Moreover, the interaction between the pRb family proteins and the E2F family transcription factors plays a central role in governing cell cycle progression and DNA replication by controlling the expression of cell cycle E2F-dependent genes. In addition, pRb recruits chromatin remodeling factors such as histone deacetylase 1 (HDAC1) SWI/SNF factors, Polycomb group proteins or methyltransferase that act on the nearby surrounding nucleosome structure.
The most convincing evidence of the importance of pRb in cellular differentiation comes from studies of Rb knockout mice, where the disruptions of the Rb gene cause death by day 14 of gestation, associated with defects in the development of the hematopoietic system and central nervous system
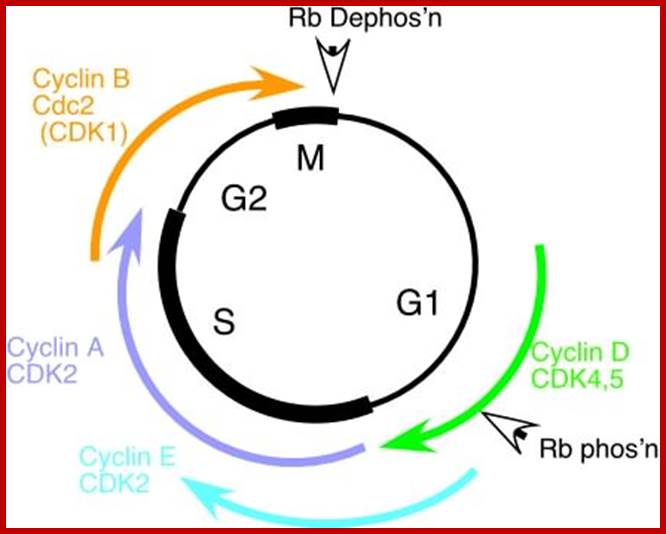
The diagram depict different stages of cell cycle regulated by different sets of Cdc-Cdks and RB proteins; fmc.med.univ-tours.fr
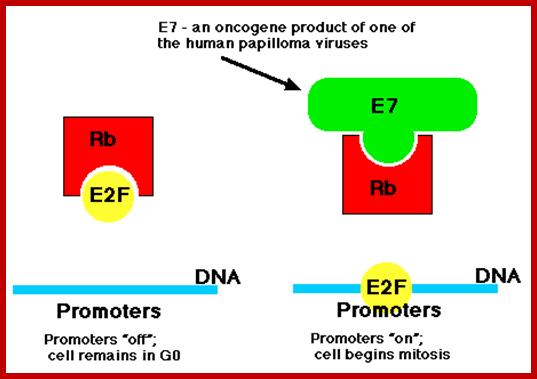
Once inside the cells of their host, these viruses synthesize
- a protein designated E7 and another designated E6.
· Of the >30 strains of HPV that infect humans, several, especially HVP-16 and HPV-18, have been implicated as a risk factor for for cervical cancer and also cancers of the throat. Their E7 protein binds to the Rb protein preventing it from binding to the host transcription factor E2F.
· E2F is now free to bind to the promoters of genes (like c-myc) that cause the cell to enter the cell cycle (right). Thus this version of E7 is an oncogene product.
· The E6 protein binds the p53 protein targeting it for destruction by proteasomes and thus removing the block on the host cell's entering the cell cycle.

There is a short window in the mammalian cell cycle during which cells can respond to extracellular cues by withdrawing temporarily from the cell cycle. When these cells re-enter the cell cycle, they require several extra hours in the G1 phase before they replicate their DNA compared with their cycling counterparts. More than 20 years after this initial observation, we still do not understand what is taking so long. Hillary A.Coller; www.nature.com
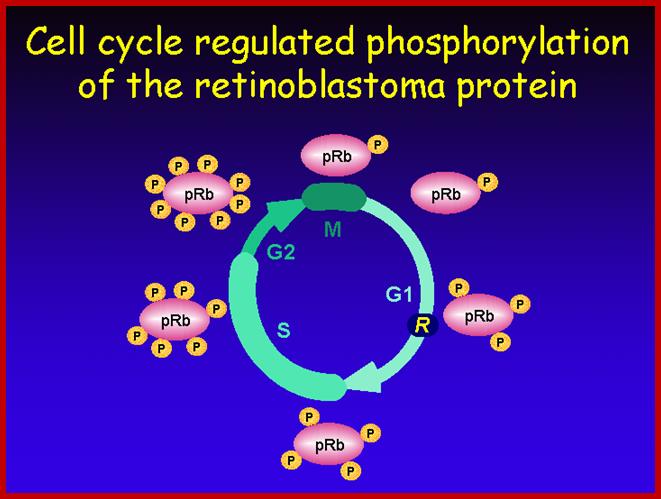
G1- Rb,P107, P130, bind to E2Fdelay to enter G1 and ; when DNA damage occurs-The rapid response involves phosphorylation events that initiate with either kinase ATM (Ataxia telangiectasia mutated) or ATR (Ataxia Telangiectasia and Rad3 related), which act as the sensors, depending on the type of damage.
- The S-phase is critical for the single stranded chromosome becomes double stranded by means of DNA replication, yet they are held together all along the length of the chromosome and also at centromeric region which has an elaborate structural organization called kinetochore (Centrosome). Each of the chromosomes contain one long DNA compacted by nucleosomal organization. Initiation of replication at origins in governed by several factors. Firing of replication is critical and it takes place only once in one cell cycle and second initiation is prevented before the M-phase is completed. During replication if there are any errors; they are fixed, and if the damage is beyond repair, the cell is subjected to Apoptosis. The progression of S-phase requires cyclin-E. Once their function is over these cyclins are degraded by SCF mediated process. The SCF complex consists of Cdc 53, SKP1 and Cdc4, all together targets Sic-1 the inhibitor of S-phase Cdk-cyclins. By ubiquitination process the Sic1 is degraded, this releases the Cdk-cyclin (G1) complex from inhibition
G2 check point- G2 stage one finds rapid growth; and DNA replicates, DNA damage triggers the activation of the aforementioned ATM/ATR pathway, in which ATM/ATR phosphorylate and activate the Chk1/Chk2 checkpoint kinases. Chk1/2 phosphorylate cdc25 which, in addition to being inhibited, is also sequestered in the cytoplasm by the 14-3-3 proteins. 14-3-3 are upregulated by p53, which, as previously mentioned, is activated by Chk1 and ATM/ATR. p53 also transactivates p21, and both p21 and the 14-3-3 in turn inhibit cyclin B-cdc2 complexes through the phosphorylation and cytoplasmic sequestering of cdc2. In addition, the inactivation of cdc25 results in its inability to dephosphorylate and activate cdc2.[12][13] Finally, another mechanism of damage response is through the negative regulation of Plk1 by ATM/ATR, which in turn results in the stabilization of Wee1 and Myt1, which can then phosphorylate and inhibit cdc2, thus keeping the cell arrested in G2 until the damage is fixed.
Following the decision to enter the cell cycle and undergo division, the cell goes through S phase, in which it replicates its DNA, and, in most species, G2, in which it undergoes rapid growth and protein synthesis in preparation for mitosis, the process of cell division. The G2/M checkpoint, also known as the DNA damage checkpoint, ensures that the cell underwent all of the necessary changes during the S and G2 phases and is ready to divide.
The primary complex responsible for the transition from G2 to M is the Cyclin B-cdc2 (CDK1 homolog) complex. The activity of cdc2 is regulated directly by cyclins B and by the phosphatase cdc25. Prior to entry into mitosis, cdc2 is maintained in an inactive state by the kinases Wee1 and Myt1, which phosphorylate tyrosine residues on cdc2. As the cell progresses through G2 and reaches the G2/M transition, the kinase Plk1 phosphorylates Wee1, which targets Wee1 for degradation via the SCF ubiquitin ligase complex.[10] An additional function of Plk1 is to activate Cdc25 through phosphorylation. The compound effect of Wee1 degradation and Cdc25 activation is the net removal of inhibitory phosphorylation from cdc2, which activates cdc2. Plk1 is activated at the G2/M transition by the Aurora A and Bora, which accumulate during G2 and form an activation complex. The Plk1-Cdc2-cdc25 complex then initiates a positive feedback loop which serves to further activate Cdc2, and in conjunction with an increase in cyclin B levels during G2, the resulting cdc2-cyclin B complexes then activate downstream targets which promote entry into mitosis.[11]
The mechanisms by which mitotic entry is prevented in response to DNA damage are similar to those in the G1/S checkpoint. DNA damage triggers the activation of the aforementioned ATM/ATR pathway, in which ATM/ATR phosphorylate and activate the Chk1/Chk2 checkpoint kinases. Chk1/2 phosphorylates cdc25 which, in addition to being inhibited, is also sequestered in the cytoplasm by the 14-3-3 proteins. 14-3-3 are upregulated by p53, which, as previously mentioned, is activated by Chk1 and ATM/ATR. p53 also transactivates p21, and both p21 and the 14-3-3 in turn inhibit cyclin B-cdc2 complexes through the phosphorylation and cytoplasmic sequestering of cdc2. In addition, the inactivation of cdc25 results in its inability to dephosphorylate and activate cdc2.[12][13] Finally, another mechanism of damage response is through the negative regulation of Plk1 by ATM/ATR, which in turn results in the stabilization of Wee1 and Myt1, which can then phosphorylate and inhibit cdc2, thus keeping the cell arrested in G2 until the damage is fixed.
The G2 stage is again a preparatory stage for M-phase, which requires a whole set of proteins and organization of cellular components for chromosomal separation and cytoplasm division. If the DNA damage is not repaired in the S-phase and even in G2 phase the cell won’t enter into M-phase. Cells have in built sensory system.
- The M phase, though short in duration, involves a major physical changes,; dismemberment of nuclear membrane and pore-complexes, dismemberment of endoplasmic reticulum, disassembly of microtubules and actin filaments to their respective subunits tubulins and actins, organization mitotic apparatus, including tractile fibers at centrosome, separation of chromatids and centromere, movement of chromatids to their respective poles and finally cytoplsmic division. These events can be visualized. Among the M-phase events the regulatory complex that plays an important role is Anaphase Promoting Complex (APC).
Mutations that exert cell cycle control in yeast are: Cdc 28 at G1-à S; Cdc 28 at S phase; cdc24 at G-2 and Cdc 34 M-à G1
Genetic analysis of single cell systems such as Saccharomyces cerevisiae (budding yeast) and Saccharaomyces pombe (fission yeast) has yielded a wealth of database information. Combined with genetic data, Proteome search has provided information about the number of genes and gene products involved in cell cycle control. The database search to 95% accuracy shows that Homo sapiens have 99 gene products [28-protein for G1/S, 28 proteins for -G2/M, 23 proteins for -M, 41-Sphase, 24-others], Mus musculus contain 68 gene products and S.cerevisiae 87 to name only few. Important factors that operate cell cycle control system are cell cycle specific kinase complexes and few cell cycle kinase inhibitors. Among many of the kinases cyclin dependent kinases (CDK) are important; for their activity cyclins are required, hence the name cyclin dependent kinases. Kinases are effector molecules and cyclins are required for kinase effector function. Perhaps the first such kinase discovered was from yeast and it was called Maturation promoting factor (MPF), later it turned out to be Mitotic Promoting Factor (MPF) or mitosis Promoting Kinase (MPK). This protein complex turned out to be a serine-threonine protein kinase but dependent on specific cyclin. From a variety of sources many such cyclin dependent kinases and cyclins have been discovered their nomenclature has not been strictly adhered to International nomenclature rules, so there is confusion in identifying, which is which.
Role of P53: In normal cells, the p53 protein level is low. DNA damage and other stress signals may trigger the increase of p53 proteins, which have three major functions: growth arrest, DNA repair and apoptosis (cell death). The growth arrest stops the progression of cell cycle, preventing replication of damaged DNA.
When DNA damage occurs, or when the cell detects any defects which necessitate it to delay or halt the cell cycle in G1, arrest occurs through several mechanisms. The rapid response involves phosphorylation events that initiate with either kinase ATM (Ataxia telangiectasia mutated) or ATR (Ataxia Telangiectasia and Rad3 related), which act as the sensors, depending on the type of damage. These kinases phosphorylate and activate the effector kinases Chk2 and Chk1, respectively, which in turn phosphorylate the phosphatase Cdc25A, thus marking it for ubiquitination and degradation.
To maintain the arrest, another response is initiated, by which Chk2 or Chk1 phosphorylate p53, a tumor suppressor, and this stabilizes p53 by preventing it from binding Mdm2, a ubiquitin ligase which inhibits p53 by targeting it for degradation. The stable p53 then acts a transcriptional activator of several target genes, including p21, an inhibitor of the G1-to-S promoting complex cyclin E-CDK21. In addition, another mechanism by which p21 is activated is through the accumulation of p16 in response to DNA damage. p16 disrupts cyclin D-CDK4 complexes, thus causing the release of p21 from the complexes, which leads to the dephosphorylation and activation of Rb, which allows Rb to bind and inhibit E2F1-3, thus keeping the cell from transitioning to S phase. Recently, some aspects of this model have been disputed.
G2- check point-Cyclin B-cdc2 (CDK1 homolog) complex- enry from G2 to M, CDC25; DNA damage triggers the activation of the aforementioned ATM/ATR pathway, in which ATM/ATR phosphorylate and activate the Chk1/Chk2 checkpoint kinases; Chk1/2 phosphorylate cdc25 which, in addition to being inhibited, is also sequestered in the cytoplasm by the 14-3-3 proteins. 14-3-3 are upregulated by p53, which, as previously mentioned, is activated by Chk1 and ATM/ATR. p53 also transactivates p21, and both p21 and the 14-3-3 in turn inhibit cyclin B-cdc2 complexes through the phosphorylation and cytoplasmic sequestering of cdc2.
M phase- The mitotic spindle checkpoint occurs at the point in metaphase where all the chromosomes should/have aligned at the mitotic plate and be under bipolar tension. The tension created by this bipolar attachment is what is sensed, which initiates the anaphase entry. To do this, the sensing mechanism ensures that the anaphase-promoting complex (APC/C) is no longer inhibited, which is now free to degrade cyclin B, which harbors a D-box (destruction box), and to break down securin. The latter is a protein whose function is to inhibit separase, which in turn cuts the cohesins, the protein composite responsible for cohesion of sister chromatids. Once this inhibitory protein is degraded via ubiquitination and subsequent proteolysis, separase then causes sister chromatid separation. After the cell has split into its two daughter cells, the cell enters G1. The G2-M DNA damage checkpoint is an important cell cycle checkpoint ineukaryotic organisms ranging from yeast to mammals
G2 stage one finds rapid growth; and DNA replicates, DNA damage triggers the activation of the aforementioned ATM/ATR pathway, in which ATM/ATR phosphorylate and activate the Chk1/Chk2 checkpoint kinases. Chk1/2 phosphorylate cdc25 which, in addition to being inhibited, is also sequestered in the cytoplasm by the 14-3-3 proteins. 14-3-3 are upregulated by p53, which, as previously mentioned, is activated by Chk1 and ATM/ATR. p53 also transactivates p21, and both p21 and the 14-3-3 in turn inhibit cyclin B-cdc2 complexes through the phosphorylation and cytoplasmic sequestering of cdc2. In addition, the inactivation of cdc25 results in its inability to dephosphorylate and activate cdc2. Finally, another mechanism of damage response is through the negative regulation of Plk1 by ATM/ATR, which in turn results in the stabilization of Wee1 and Myt1, which can then phosphorylate and inhibit cdc2, thus keeping the cell arrested in G2 until the damage is fixed.
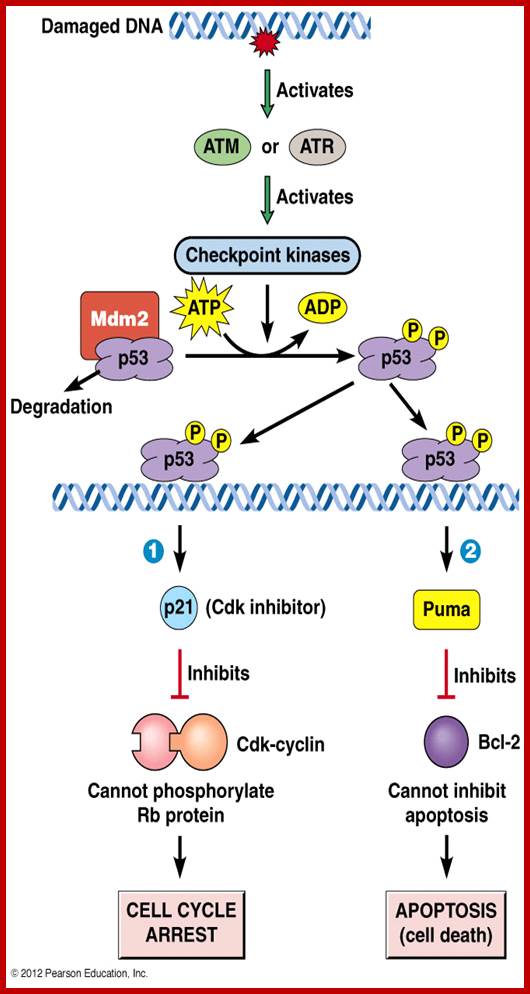
DNA maintenance-Checkpoint
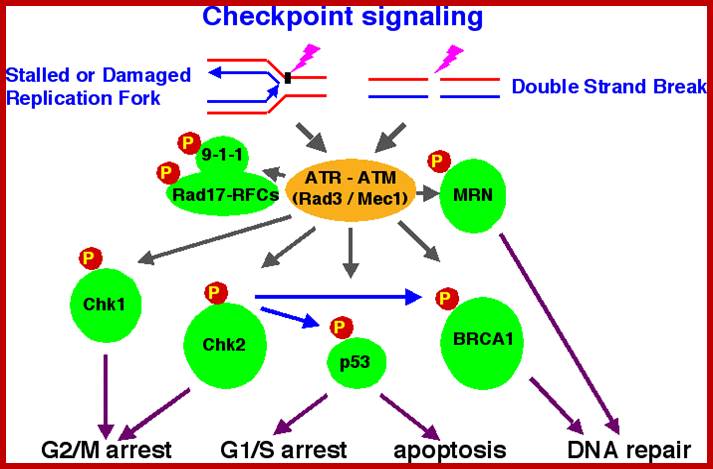
DNA replication checkpoint
Accurate replication of the millions or billions of DNA base pairs in a eukaryotic genome is a remarkable achievement. This accomplishment is even more astonishing when one considers for DNA synthesis are rarely ideal. Damaged template, protein complexes bound to DNA, and poor supply of dNTPs are among the many obstacles that must be overcome to replicate genome. All of these situations can stall replication forks. Stalled forks pose grave threats to genome integritybecause they can rearrange, break, or collapse through disassembly of the replication complex 5. The pathways that respond to replication stress are signal transduction pathways that are conserved across evolution 6 7. Atop the pathways are also ATM/ATR family kinases. These kinases together with a trimeric checkpoint clamp (termed 9-1-1 complex) and five-subunit checkpoint clamp loader (Rad17-RFC2-RFC3-RFC4-RFC5) senses stalled replication forks and transmit a checkpoint signal3. One of major functions of replication checkpoint is to prevent the onset of mitosis by regulating mitotic control proteins such as Cdc25. But perhaps the most important activity of replication checkpoint is to stabilize and protect replication forks 8. The protein kinase Cds1 (human Chk2homolog; in human, Chk1 is a functional Cds1 homolog) is a critical effector of the replication checkpoint in the fission yeast Schizosaccharomycs pombe , Cds1 is required to prevent stabilization of replication fork in cells treated with hydroxyurea (HU), a ribonucleotide reductase inhibitor that stalls replication by depleting dNTPs 11. In the budding yeast Saccharomyces cerevisiae, a failure to activate Rad53 (Chk2 homolog) is associated with collapse and regression of replication forks and gross chromosomal rearrangements in cells treated with HU
Accurate duplication of eukaryotic genome is a challenging task, given that environment of cell growth and division is rarely ideal. Cells are constantly under the stress of intrinsic and extrinsic agents that cause DNA damage or interference with DNA replication. To cope with these assaults, cells are equipped with DNA maintenance check points to arrest cell cycle and facilitate DNA repair pathways. DNA maintenance checkpoints include (a) the DNA damage checkpoints that recognize and respond to DNA damage, and (b) the DNA replication checkpoint that monitors the fidelity of copying DNA.
Damage checkpoint;
DNA Replication fork protection complex (FPC)

The DNA replication checkpoint stabilizes replication forks that have stalled at DNA adducts and other lesions that block DNA polymerases. In the absence of DNA replication checkpoint, stalled forks are thought to collapse, creating strand break that threatens genome stability and cell viability. Therefore, discovering how cells cope with aberrant replication forks is essential for understanding mechanisms of genome maintenance. The Chk1 and Chk2/Cds1 checkpoint kinases, which are key mediators of DNA damage and DNA replication checkpoints, are thought to be involved in cancer development. We found the Swi1 protein is required for survival of replication fork arrest and effective activation of Chk2 kinase in fission yeast. Swi1 forms tight complex with Swi3 protein and moves with replication forks. Swi1-Swi3 complex is also important for proficient DNA replication even in the absence of agents that cause genotoxic stress, creating single-strand DNA gaps at replication forks. These results led us to propose Swi1-Swi3 define a replication fork protection complex (FPC) that stabilizes replication forks in a configuration that is recognized by replication checkpoint sensors. Interestingly, Tof1protein (Budding yeast Swi1 homolog) has been reported to have similar functions. Tof1 is also involved in Rad53 (Chk2 homolog) activation and travels with replication fork. Tof1 is needed to restrain fork progression when DNA synthesis is inhibited by HU indicating that Tof1 is required for coordination of DNA synthesis and replisome (replication machinery) movement
DNA damage checkpoints are classified into at least 3 checkpoints: G1/S (G1) checkpoint, intra-S phase checkpoint, and G2/M checkpoint. DNA replication checkpoint that arrests cell cycle at G2/M transition until DNA replication is complete. There are more checkpoints such as Spindle checkpoint and Morphogenesis checkpoint
Checkpoints are regulatory mechanisms that block cell cycle progression when key cellular processes are defective or chromosomes are damaged. During meiosis, genetic recombination between homologous chromosomes is essential for proper chromosome segregation at the first meiotic division. In response to incomplete recombination, the pachytene checkpoint (also known as the meiotic recombination checkpoint) arrests or delays meiotic cell cycle progression, thus preventing the formation of defective gametes. http://jcs.biologists.org/
Here, we describe a role for a meiosis-specific kinase, Mek1, in the meiotic recombination checkpoint in fission yeast. Mek1 belongs to the Cds1/Rad53/Chk2 family of kinases containing fork head-associated domains, which participate in a number of checkpoint responses from yeast to mammals.
During the meiotic cell cycle, a surveillance mechanism called the “pachytene checkpoint” ensures proper chromosome segregation by preventing meiotic progression when recombination and chromosome synapsis are defective. The silencing protein Dot1 (also known as Pch1) is required for checkpoint-mediated pachytene arrest of thezip1 and dmc1 mutants of Saccharomyces cerevisiae. In the absence of DOT1,(Pch1) the zip1and dmc1 mutants inappropriately progress through meiosis, generating inviable meiotic products. http://www.ncbi.nlm.nih.gov/
Other components of the pachytene checkpoint include the nucleolar protein Pch2 and the heterochromatin component Sir2. In dot1, disruption of the checkpoint correlates with the loss of concentration of Pch2 and Sir2 in the nucleolus.
In Saccharomyces cerevisiae, several components of the DNA damage checkpoint have been identified. Rad9 and the Rad24 group of proteins (Rad24, Rad17, Mec3, and Ddc1) are thought to be involved in sensing damage and/or generating a signal in response to damage.
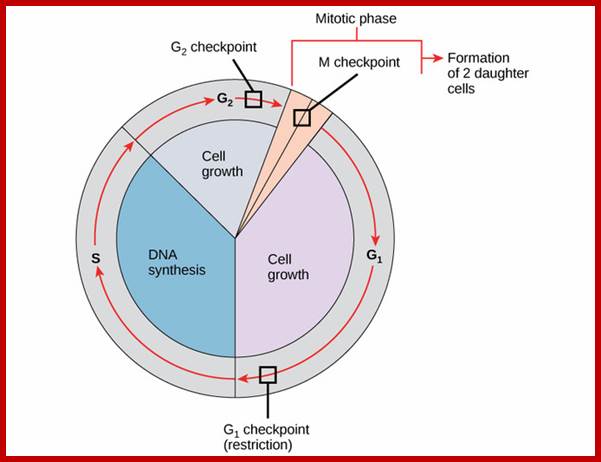
Internal check points during cell cycle.
Saccharomyces cerevisiae zip1 mutant, which exhibits defects in synaptonemal complex formation and meiotic recombination, triggers a checkpoint that causes cells to arrest at the pachytene stage of meiotic prophase. These results suggest that meiotic chromosomal proteins function in the signaling of meiotic prophase defects and that the correct stoichiometry of Red1, Mek1, and Hop1 is needed to achieve checkpoint-mediated cell cycle arrest at pachytene. http://mcb.asm.org/;
However, in cells in which the function of the DNA-replication-checkpoint . It is the DSBs in the HU-treated mutant cells occurred at normal sites and were associated with recombination. In addition, Cdc2p is apparently not involved in this process. We propose that the sequence of meiotic S phase and initiation of recombination is coordinated by DNA-replication-checkpoint proteins. http://www.pnas.org/
Spindle assembly check point prevents chromosome mis-segregation bot at mitosis as weel as meiosis. Defects in spindle assembly check points are unlikely to cause cancer ?
Meiotic check points protein kinases CHEK1 And Check2; The central role in meiosis of human and mouse CHEK1 and CHEK2 and their orthologs in Saccharomyces cerevisiae,Caenorhabditis elegans, Schizosaccharomyces pombe and Drosophila has been reviewed by MacQueen and Hochwagen and Subramanian and Hochwagen. During meiotic recombination in human and mouse, CHEK1protein kinase is important for integrating DNA damage repair with cell cycle arrest. CHEK1 is expressed in the testes and associates with meiotic synaptonemal complexes during the zygonema and pachynema stages. CHEK1 likely acts as an integrator for ATM and ATR signals and in monitoring meiotic recombination.[12] In mouse oocytes CHEK1 appears to be indispensable for prophase I arrest and to function at the G2/M checkpoint.
CHEK2 regulates cell cycle progression and spindle assembly during mouse oocyte maturation and early embryo development. Although CHEK2 is a down stream effector of the ATM kinase that responds primarily to double-strand breaks it can also be activated by ATR (ataxia-telangiectasia and Rad3 related) kinase that responds primarily to single-strand breaks. In mouse, CHEK2 is essential for DNA damage surveillance in female meiosis. The response of oocytes to DNA double-strand break damage involves a pathway hierarchy in which ATR kinase signals to CHEK2 which then activates p53 and p63 proteins.
The Saccharomyces cerevisiae zip1 mutant, which exhibits defects in synaptonemal complex formation and meiotic recombination, triggers a checkpoint that causes cells to arrest at the pachytene stage of meiotic prophase. Imbalance of meiotic chromosomal proteins inactivates the pachytene checkpoint. Overproduction of Red1 or Mek1 specifically promotes sporulation of mutants that normally undergo checkpoint-mediated arrest at pachytene. Thezip1, zip2, dmc1, and hop2 mutants all exhibit defects in both recombination and synapsis; however, the molecular signal that triggers arrest in these strains remains unknown. http://mcb.asm.org/
A model of functions of the checkpoint clamp and clamp loader in crossover formation;
Four possible pathways of meiotic recombination are shown. Rad51 and Dmc1 cooperate in pathways that display interhomolog bias (three leftmost pathways). Together with Rad51–Dmc1, the 9‐1‐1 clamp and Mec1 suppress inter-sister and ectopic recombination (right pathway). ZMM proteins specifically promote the ‘crossover‐only’ pathway (third pathway from the left) by processing interhomolog joint molecules – this depends on Hop1–Red1–Mek1. The 9‐1‐1 clamp, but not Mec1, facilitates ZMM function on the crossover‐only pathway. ZMM proteins might also promote inter-sister joint molecule formation. Non‐crossover formation is independent of ZMM proteins (the leftmost pathway). Even in the absence of ZMM function, interhomolog joint molecules are formed (the second pathway from the left). In this pathway, joint molecules are resolved into either crossover or NCO.
The regulation of Cdc25C is crucial for the operation of the DNA-damage checkpoint. Two protein kinases, Chk1 and Cds1, can be activated in response to DNA damage or in the presence of unreplicated DNA. Chk1 and Cds1 may phosphorylate Cdc25C to prevent entry into mitosis through inhibition of Cdc2 (Cdk1) dephosphorylation. http://link.springer.com/
DNA damage triggers the activation of the, afore mentioned ATM/ATR pathway, in which ATM/ATR phosphorylate and activate the Chk1/Chk2 checkpoint kinases. Chk1/2 phosphorylate cdc25 which, in addition to being inhibited, is also sequestered in the cytoplasm by the 14-3-3 proteins. 14-3-3 are upregulated by p53, which, as previously mentioned, is activated by Chk1 and ATM/ATR. p53 also transactivates p21, and both p21 and the 14-3-3 in turn inhibit cyclin B-cdc2 complexes through the phosphorylation and cytoplasmic sequestering of cdc2.
Metaphase check point: Mitotic spindle check point- in metaphase- all chromosomes aligned at the middle region- bipolar attachment. To do this, the sensing mechanism ensures that the APC/C is no longer inhibited which is now free to degrade cyclin B, which harbors a D-box (destruction box), and to break down Securin. The latter is a protein whose function is to inhibit separase, which in turn cuts the Cohesins, the protein composite responsible for cohesion of sister chromatids.[16] Once this inhibitory protein is degraded via ubiquitination and subsequent proteolysis, separase then causes sister chromatid separation.[17] After the cell has split into its two daughter cells, the cell enters G1. https://en.wikipedia.org/wiki/
The tension created by this bipolar attachment is what is sensed, which initiates the anaphase entry. To do this, the sensing mechanism ensures that the anaphase-promoting complex (APC/C) is no longer inhibited, which is now free to degrade cyclin B, which harbors a D-box (destruction box), and to break down Securin. The latter is a protein whose function is to inhibit separase, which in turn cuts the Cohesins, the protein composite responsible for cohesion of sister chromatids. Once this inhibitory protein is degraded via ubiquitination and subsequent proteolysis, separase then causes sister chromatid separation.[17] After the cell has split into its two daughter cells, the cell enters G1.
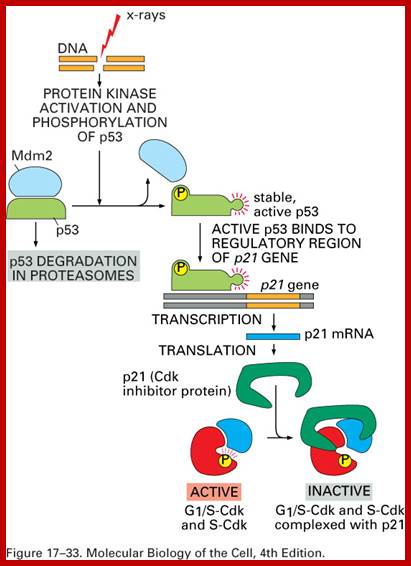
http://www.pha.jhu.edu/~ghzheng
When DNA damage occurs, or when the cell detects any defects which necessitate it to delay or halt the cell cycle in G1, arrest occurs through several mechanisms. The rapid response involves phosphorylation events that initiate with either kinase ATM (Ataxia telangiectasia mutated) or ATR (Ataxia Telangiectasia and Rad3 related), which act as the sensors, depending on the type of damage. These kinases phosphorylate and activate the effector kinases Chk2 and Chk1, respectively, which in turn phosphorylate the phosphatase Cdc25A, thus marking it for ubiquitination and degradation.
Following the decision to enter the cell cycle and undergo division, the cell goes through S phase, in which it replicates its DNA, and, in most species, G2, in which it undergoes rapid growth and protein synthesis in preparation for mitosis, the process of cell division. The G2/M checkpoint, also known as the DNA damage checkpoint, ensures that the cell underwent all of the necessary changes during the S and G2 phases and is ready to divide.
The primary complex responsible for the transition from G2 to M is the Cyclin B-cdc2 (CDK1 homolog) complex. The activity of cdc2 is regulated directly by cyclins B and by the phosphatase cdc25. Prior to entry into mitosis, cdc2 is maintained in an inactive state by the kinases Wee1 and Myt1, which phosphorylate tyrosine residues on cdc2. As the cell progresses through G2 and reaches the G2/M transition, the kinase Plk1 phosphorylates Wee1, which targets Wee1 for degradation via the SCF ubiquitin ligase complex. An additional function of Plk1 is to activate Cdc25 through phosphorylation. The compound effect of Wee1 degradation and Cdc25 activation is the net removal of inhibitory phosphorylation from cdc2, which activates cdc2. Plk1 is activated at the G2/M transition by the Aurora A and Bora, which accumulate during G2 and form an activation complex. The Plk1-Cdc2-cdc25 complex then initiates a positive feedback loop which serves to further activate Cdc2, and in conjunction with an increase in cyclin B levels during G2, the resulting cdc2-cyclin B complexes then activate downstream targets which promote entry into mitosis.
The mechanisms by which mitotic entry is prevented in response to DNA damage are similar to those in the G1/S checkpoint. DNA damage triggers the activation of the aforementioned ATM/ATR pathway, in which ATM/ATR phosphorylate and activate the Chk1/Chk2 checkpoint kinases. Chk1/2 phosphorylate cdc25 which, in addition to being inhibited, is also sequestered in the cytoplasm by the 14-3-3 proteins. 14-3-3 are upregulated by p53, which, as previously mentioned, is activated by Chk1 and ATM/ATR. p53 also transactivates p21, and both p21 and the 14-3-3 in turn inhibit cyclin B-cdc2 complexes through the phosphorylation and cytoplasmic sequestering of cdc2. In addition, the inactivation of cdc25 results in its inability to dephosphorylate and activate cdc2. Finally, another mechanism of damage response is through the negative regulation of Plk1 by ATM/ATR, which in turn results in the stabilization of Wee1 and Myt1, which can then phosphorylate and inhibit cdc2, thus keeping the cell arrested in G2 until the damage is fixed.
G2 stage one finds rapid growth; and DNA replicates, DNA damage triggers the activation of the aforementioned ATM/ATR pathway, in which ATM/ATR phosphorylate and activate the Chk1/Chk2 checkpoint kinases. Chk1/2 phosphorylate cdc25 which, in addition to being inhibited, is also sequestered in the cytoplasm by the 14-3-3 proteins. 14-3-3 are upregulated by p53, which, as previously mentioned, is activated by Chk1 and ATM/ATR. p53 also transactivates p21, and both p21 and the 14-3-3 in turn inhibit cyclin B-cdc2 complexes through the phosphorylation and cytoplasmic sequestering of cdc2. In addition, the inactivation of cdc25 results in its inability to dephosphorylate and activate cdc2. Finally, another mechanism of damage response is through the negative regulation of Plk1 by ATM/ATR, which in turn results in the stabilization of Wee1 and Myt1, which can then phosphorylate and inhibit cdc2, thus keeping the cell arrested in G2 until the damage is fixed.
The G2/M checkpoint prevents cells containing damaged DNA from entering mitosis (M). Activated CDK1 (cdc2) bound to cyclin B promotes entry into M-phase. Wee1 and Myt1 kinases and cdc25 phosphatase competitively regulate CDK1 activity; Wee1 and Myt1 inhibit CDK1 and prevent entry into M-phase, while cdc25 removes inhibitory phosphates. DNA damage activates multiple kinases that phosphorylate kinases Chk1/2 and tumor suppressor protein p53. Chk1/2 kinases stimulate Wee1 activity and inhibit cdc25C, preventing entry into M-phase. Phosphorylation of p53 promotes dissociation between p53 and MDM2 and allows binding of the transcription factor to DNA.
Pathway Description:
The G2/M DNA damage checkpoint serves to prevent the cell from entering mitosis (M-phase) with genomic DNA damage. Specifically, the activity of the Cyclin B-cdc2 (CDK1) complex is pivotal in regulating the G2-phase transition wherein cdc2 is maintained in an inactive state by the tyrosine kinases Wee1 and Myt1. It is thought that coordinated action of the kinase Aurora A and the cofactor Bora activate PLK1 as cells approach the M-phase, which in turn activates the phosphatase cdc25 and downstream cdc2 activity, hence establishing a feedback amplification loop that efficiently drives the cell into mitosis. Importantly, DNA damage cues activate the sensory DNA-PK/ATM/ATR kinases, which relay two parallel cascades that ultimately serve to inactivate the Cyclin B-cdc2 complex. The first cascade rapidly inhibits progression into mitosis: the Chk kinases phosphory- late and inactivate cdc25, which prevents activation of cdc2. The slower second parallel cascade involves phosphorylation of p53 and allows for its dissociation from MDM2 and MDM4 (MdmX), which activates DNA binding and transcriptional regulatory activity, respectively. The transcriptional ability of p53 is further augmented through acetylation by the co-activator complex p300/PCAF. The second cascade constitutes the p53 downstream-regulated genes including: 14-3-3, which binds to the phosphorylated Cyclin B-cdc2 complex and exports it from the nucleus; GADD45, which binds to and dissociates the Cyclin B-cdc2 complex; and p21 Cip1, an inhibitor of a subset of the cyclin-dependent kinases including cdc2. Recent data suggest an important role for the p53-regulated WIP1 phosphatase that acts as a critical dampener of DNA damage signalling in cancer. In human cancer, researchers have found p53 to be commonly mutated, indicating that this checkpoint is a critical barrier to tumour formation. In addition, sporadic and familial mutations in the DNA-repair proteins such as the BRCA-family, ATM, and the Fanconi Anaemia proteins further highlight this as a key tumour suppressor checkpoint.
G2-M A commentary on the G2/M transition of the plant cell cycle;
Background The complex events of mitosis rely on precise timing and on immaculate preparation for their success, but the G2/M transition in the plant cell cycle is currently steeped in controversy and alternative models.
Scope In this brief review, the regulation of the G2/M transition in plants is commented on. The extent to which the G2/M transition is phosphoregulated by WEE1 kinase and CDC25 phosphatase, as exemplified in yeasts and animals, is discussed together with an alternative model that excludes these proteins from this transition. Arabidopsis T-DNA insertional lines for WEE1 and CDC25 that develop normally prompted the latter model. An argument is then presented that environmental stress is the norm for higher plants in temperate conditions. If so, the repressive role that WEE1 has under checkpoint conditions might be part of the normal cell cycle for many proliferative plant cells. Arabidopsis CDC25 can function as either a phosphatase or an arsenate reductase and recent evidence suggests that cdc25 knockouts are hypersensitive to hydroxyurea, a drug that induces the DNA-replication checkpoint. That other data show, a null response of these knockouts to hydroxyurea leads to an airing of the controversy surrounding the enigmatic plant CDC25 at the G2/M transition. Dennis Francis; http://aob.oxfordjournals.org/https://www.cellsignal.com/
Metaphase check point; The tension created by this bipolar attachment is what is sensed, which initiates the anaphase entry. To do this, the sensing mechanism ensures that the anaphase-promoting complex (APC/C) is no longer inhibited, which is now free to degrade cyclin B, which harbors a D-box (destruction box), and to break down Securin. The latter is a protein whose function is to inhibit separase, which in turn cuts the Cohesins, the protein composite responsible for cohesion of sister chromatids. Once the inhibitory protein is degraded via ubiquitination and subsequent proteolysis, separase then causes sister chromatid separation. After the cell has split into its two daughter cells, the cell enters G1.
The mitotic spindle checkpoint occurs at the point in metaphase where all the chromosomes should/have aligned at the mitotic plate and be under bipolar tension. The tension created by this bipolar attachment is what is sensed, which initiates the anaphase entry. To do this, the sensing mechanism ensures that the anaphase-promoting complex (APC/C) is no longer inhibited, which is now free to degrade cyclin B, which harbors a D-box (destruction box), and to break down Securin. The latter is a protein whose function is to inhibit separase, which in turn cuts the Cohesins, the protein composite responsible for cohesion of sister chromatids. Once this inhibitory protein is degraded via ubiquitination and subsequent proteolysis, separase then causes sister chromatid separation. After the cell has split into its two daughter cells, the cell enters G1.
Spindle assembly checkpoint;
Spindle assembly prevents the advancement into anaphase.
The mechanism responsible for ensuring appropriate spindle assembly is called the spindle assembly checkpoint.
If any one kinetochore of either sister chromatid does
not appropriately attach to spindle spindle
microtubule, then transition to anaphase is inhibited.
It has been recently understood that the protein Mad2 is involved in this process. Mad2 attaches to the kinetochores that have failed to attach to microtubules and becomes activated.
It interacts with the protein Cdc20 to inhibit Cdc20activity. Because Cdc20 is an activator of APC/Cthatis required to initiate anaphase, transition to anaphase will not occur as long as activated Mad2 is present. Production of activated Mad2 will stop only when all kinetochores are properly attached to microtubules.
Thus, the spindle assembly checkpoint ensures that celldivision does not progress any further until all chromosomes are properly attached to microtubules so that the genome of the parent cell can be accurately segregated to the 2 daughter cells.
Chromosome segregation checkpoint:
Once the chromosomes have segregated properly, telophase starts. The
G2/M cyclin-CDK complex needs to be inactivated to allow transition to telophase and
for
the subsequent cytokinesis events. This checkpoint monitors thepositions of
the segregating daughter chromosomes. After proper chromosome
segregation is confirmed, the regulatory
factor Cdc14 inactivates the G2/M
cyclin-CDK complex, allowing cells to transition to telophase and
then to cytokinesis.

https://www.studyblue.com
Structure of the CHEK1 protein: Based on PyMOL rendering of PDB; Checkpoint kinase 1, commonly referred to as Chk1 is an Serine/threonine-specific protein kinase that in humans, is encoded by the CHEK1 gene. Chk1 coordinates the DNA damage response (DDR) and cell cycle checkpoint response. Activation of Chk1 results in the initiation of cell cycle checkpoints, cell cycle arrest, DNA repair and cell death to prevent damaged cells from progressing through the cell cycle. Chk1 contains four Ser/Gln residues.[6] Chk 1 activation occurs primarily through the phosphorylation of the conserved sites, Ser-317, Ser-345 and less often at Ser-366.
CHK1 protein- is serine-threonine kinase; Chk1 coordinates the DNA damage response (DDR) and cell cycle checkpoint response. Activation of Chk1 results in the initiation of cell cycle checkpoints, cell cycle arrest, DNA repair and cell death to prevent damaged cells from progressing through the cell cycle; https://en.wikipedia.org
CHK1- DNA repair, cell cycle arrest; CHK1 is activated by ATR, CHK1 regulates M phase; G2-M transition, S- phase. The protein encoded by this gene belongs to the Ser/Thr protein kinase family. It is required for checkpoint mediated cell cycle arrest in response to DNA damage or the presence of unreplicated DNA. This protein acts to integrate signals from ATM and ATR, two cell cycle proteins involved in DNA damage responses, that also associate with chromatin in meiotic prophase I. Phosphorylation of CDC25A protein phosphatase by this protein is required for cells to delay cell cycle progression in response to double-strand DNA breaks. Several alternatively spliced transcript variants have been found for this gene. [provided by Ref Seq, Oct 2011]; CHEK1 (Checkpoint Kinase 1) codes for a protein which is a kinase. Diseases associated with CHEK1 include Ataxia-telangiectasia and breast cancer. Among its related pathways are Gene Expression and Signaling by GPCR. GO annotations related to this gene include transferase activity, transferring phosphorus-containing groups and protein tyrosine kinase activity. An important paralog of this gene is SBK2.
CHK2 protein kinase:
CHK2 operates in an intricate network of proteins to elicit DNA repair, cell cycle arrest or apoptosis in response to DNA damage. Mutations to the CHEK2 gene have been linked to a wide range of cancers including breast cancer.
CHK2 operates in an intricate network of proteins to elicit DNA repair, cell cycle arrest orapoptosis in response to DNA damage. Mutations to the CHEK2 gene have been linked to a wide range of cancers including breast cancer.
The serine/threonine kinase CHK2 is a key component of the DNA damage response. In human cells, following genotoxic stress, CHK2 is activated and phosphorylates >20 proteins to induce the appropriate cellular response, which, depending on the extent of damage, the cell type, and other factors, could be cell cycle checkpoint activation, induction of apoptosis or senescence, DNA repair, or tolerance of the damage. Recently, CHK2 has also been found to have cellular functions independent of the presence of nuclear DNA lesions. In particular, CHK2 participates in several molecular processes involved in DNA structure modification and cell cycle progression. In this review, we discuss the activity of CHK2 in response to DNA damage and in the maintenance of the biological functions in unstressed cells. These activities are also considered in relation to a possible role of CHK2 in tumorigenesis and, as a consequence, as a target of cancer therapy. CHK2 kinase in the DNA damage response and beyond http://jmcb.oxfordjournals.org/
A representative crystal structure of the Chk2 protein after computational modelling and simulation of molecular dynamics. The coloured spheres represent the locations of mutations, which BII scientists have found to be useful as a prognostic marker for HG-SOC.
From 50000 to 500000 insults to the nuclear DNA of each cell of our body every day,is a the most dangerous feature of cell cycle. This happens as a result of normal metabolism. This number is further increased by the genotoxic effects of air pollution, cigarette smoking, food additives, toxins, solar ultraviolet radiation, X-ray exams, and nuclear plant disasters. Indeed, free oxygen radicals that arise during metabolism or exposure to ionizing radiation (IR) can break the phosphodiester bonds in the backbone of the DNA helix. Similarly, alkylating agents and ultraviolet irradiation can distort the DNA helix and lead to chromosomal breakage. When the lesion damages only one of the two strands of the double helix, a single strand break occurs. When two of these breaks are close on opposite strands, a double-strands can break (DSB) can and fuse with genetic material. For this reason and for the intensity of the cellular response to them, DSBs are widely studied. (Ciccia and Elledge, 2010)
From 50000 to 500000 insults to the nuclear DNA of each cell of our body every day,is a the most dangerous feature of cell cycle. This happens as a result of normal metabolism. This number is further increased by the genotoxic effects of air pollution, cigarette smoking, food additives, toxins, solar ultraviolet radiation, X-ray exams, and nuclear plant disasters. Indeed, free oxygen radicals that arise during metabolism or exposure to ionizing radiation (IR) can break the phosphodiester bonds in the backbone of the DNA helix. Similarly, alkylating agents and ultraviolet irradiation can distort the DNA helix and lead to chromosomal breakage. When the lesion damages only one of the two strands of the double helix, a single strand break occurs. When two of these breaks are close on opposite strands, a double-strands can break (DSB) can and fuse with genetic material. For this reason and for the intensity of the cellular response to them, DSBs are widely studied. (Ciccia and Elledge, 2010)
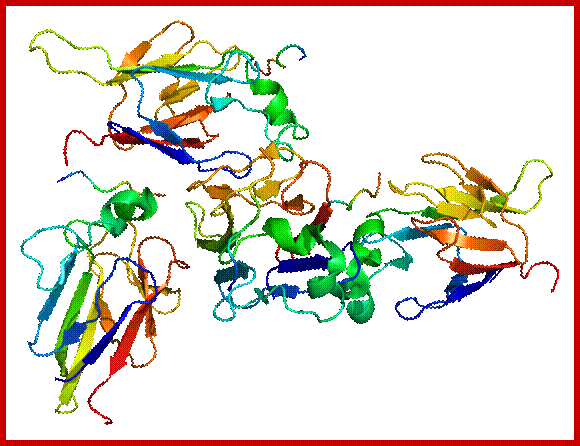
CHEK proteins are Cheek point serine/threonine kinases; CHEK1 and CHEK2 are tumor suppressor; CHK2 operates in an intricate network of proteins to elicit DNA repair, cell cycle arrest or apoptosis in response to DNA damage. Mutations to the CHEK2 gene have been linked to a wide range of cancers including breast cancer. https://en.wikipadia.org/wiki.
Additional Information-Notes:
What happens to cells that "decide" to go for cell division?
If the decision is to divide (e.g., if a growth factor signal reaches cell), then the cell will go into S phase, and DNA synthesis will begin. Once this happens, the cell must go through the cell division process, all the way through to the end. However, each stage of the cell cycle must be successfully completed, before the next stage can be entered, and each step is carefully regulated. The cell has various mechanisms, called check points, that allow it to proceed from one stage to the next only after the previous one has been confirmed to be completed. http://eishinoguchi.com.
Cell cycle control molecules were first
discovered through cell fusion experiments in
the 1970s.
The fusion of cells in different stages of the
cell cycle (to form a heterokaryon) demonstrated that latter stages possess factor
trigger progression.
G1 + S -> G1 nucleus enters S phase immediately.
S + G2 -> G2 nucleus does not enter S phase before
mitosis.
M+ G1, S or G2 -> non-M phase nuclei enters mitosis, whether or
not the DNA is duplicated.http://www.mun.ca/biology/desmid/brian/BIOL2060/BIOL2060-19/19_32.jpg
- Each of these phases are regulated by specific molecular events and factors, until each of the events in each of the phases are completed cell won’t enter into the next stage. Thus, the entry and exit to and out of stages or phases is tightly controlled by a variety of factors; and the factors themselves undergo changes in their turn over (synthesis and degradation), activation and inactivation of them.
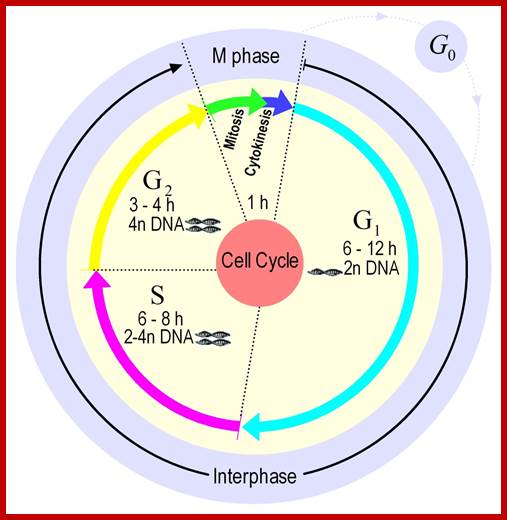
http://users.minet.uni-jena.de/
Duration required for each of the phases; The growth of all organisms requires that the genome is accurately replicated and equally partitioned between two cellular progenies. In eukaryotes, the duplication of chromosomes, the separation of sister chromatids, and their segregation to opposite poles of the cell prior to cytokinesis are features of the cell cycle and grant maintenance of genomic integrity. Eukaryotic cells have evolved a surveillance mechanism for DNA segregation, the Mitotic Spindle Assembly Checkpoint (MSAC). This checkpoint blocks anaphase onset and prevents exit from mitosis until all chromosomes are properly attached and have aligned on the mitotic spindle. Its malfunction leads to cell death, generates aneuploidy, might facilitate tumorigenesis and aging, and might contribute to cancer.
The research focuses on modeling and simulation of the human's mitosis transition controls, the MSAC and the mitotic exit. Our modeling approaches base on biochemical reaction networks. We use In particular: Differential equations, stochastic simulation, evolutionary optimization, and three dimensional effects. http://users.minet.uni-jena.de/
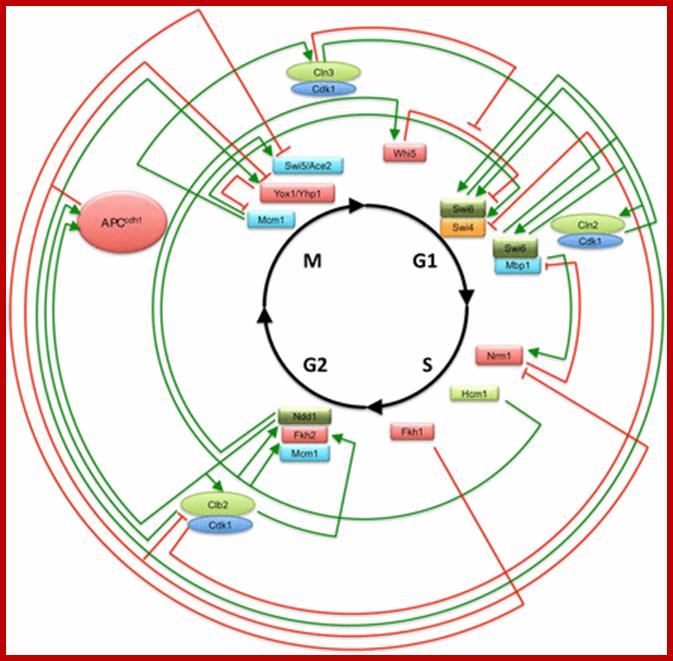
The cell-cycle transcriptional circuitry; This transcriptional circuit depicts the major interactions between transcriptional activators and repressors and their regulation by cyclin/CDK and APC discussed in the course of this article. This circuit is not meant to be exhaustive but rather to provide a reference for the interaction between the cluster regulators that are depicted in the subsequent figures. Many other interactions are discussed in the body of the article. Subunits in green are those with activating activity, subunits in red are those with repressing or inhibitory activity, and subunits in blue represent those that require a regulatory subunit. Arrows in green represent activating activities, those in red represent repressing activities, and those in black represent transitions in the process. http://www.genetics.org/
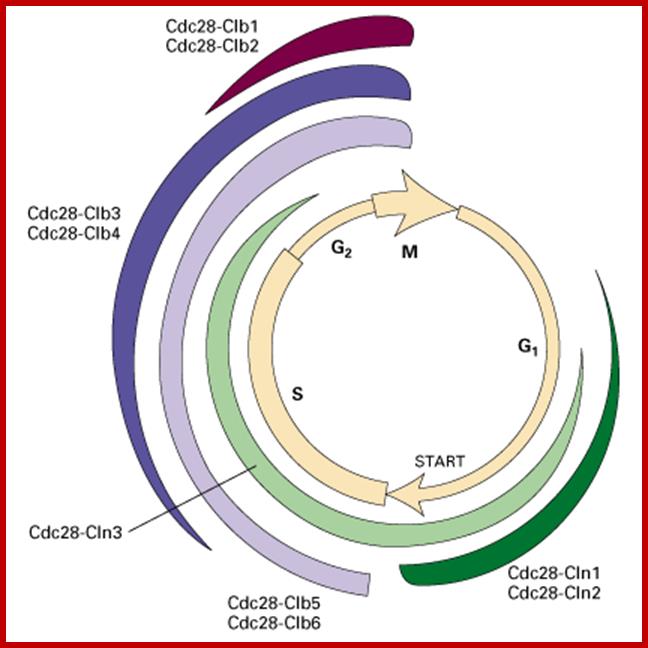
Phases at which specific cyclins and specific CDKs act; http://www.pha.jhu.edu/
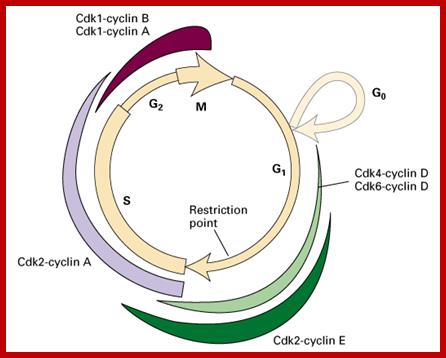
Progress of cell cycle is controlled by many cellular proteins and specifically CDKs and specific cyclins: Phases at which specific cyclins and specific CDKs act; http://www.pha.jhu.edu/
The major checkpoints lie in between G1 and S phase and G2 and M-phase and another control point exists within the M-phase events at anaphase. Notwithstanding the said checkpoints, DNA damage can introduce a checkpoint, where until the damage is repaired, cell does not enter M-phase, this can happen at S-phase or at G2 phase; if the damage is beyond repair the cell is signaled for Apoptosis.
Decide-to divide or not to divide;

To be or not to be- decide to divide or not to divide; Quiescent state-non dividing state called Go, the decision not to divide is due to not receiving growth factors such as epidermal factors or mitogens, ex. PDGF, EGF and others, but such factors are required for cells to get activated for cell division. www.oregonstate.unversity.edu
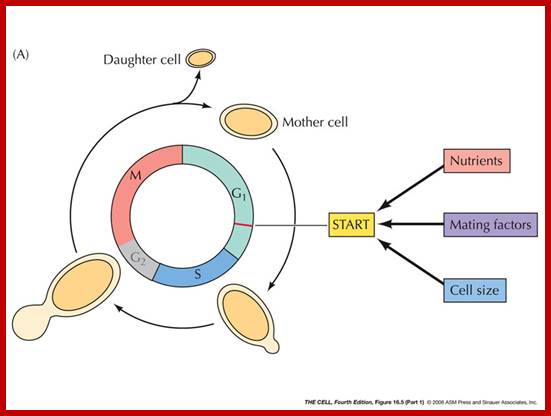
http://oregonstate.edu/
Cell Surface receptors:
Extra cellular signaling molecules have important roles in cellular development and function. For example, platelet derived growth factor-a (PDGF-a) has an important role to play in the health of the cell. Similarly, Tissue transforming growth factor-b (TGF-b has the ability to inhibit cellular proliferation of many types of cells including skin, epithelial and immune cells.
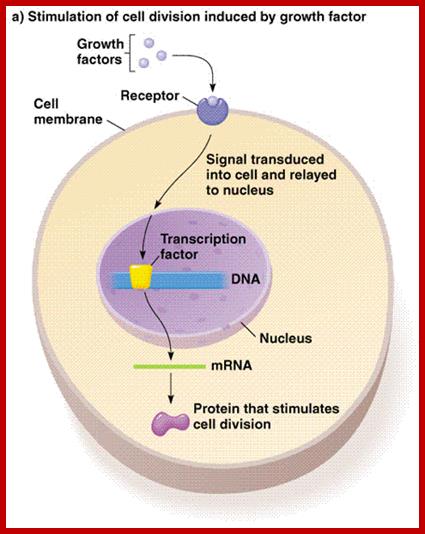
Mitogenic Growth factors binding to cell surface receptors activate MPKinases which induce cell division; http://dpuadweb.depauw.edu/
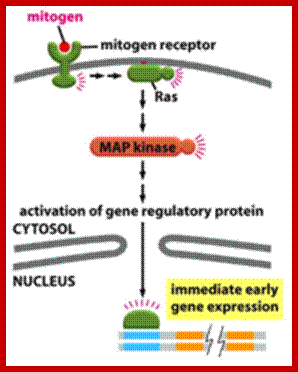
Mitogens bind to cell surface receptor, which activates cell division programme. https://quizlet.com/
There are a variety of cell surface receptors, when they bind to specific factors they get activated and it leads to activation of cell division; http://www.nature.com/
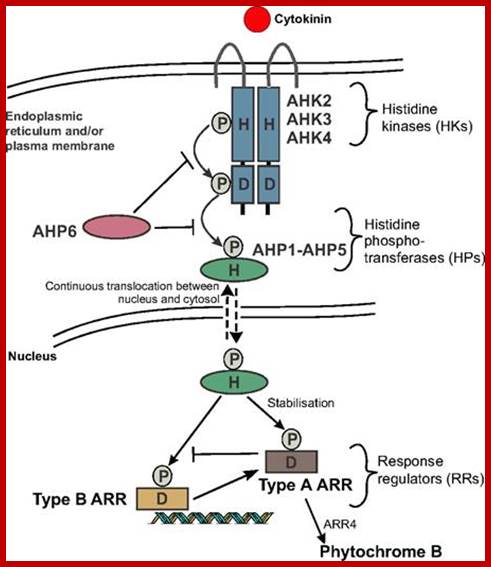
Cytokinin binds to cell surface transmembrane receptor and induce cell division via histidine phosphorylation but also induces cell differentiation; http://molbio.mgh.harvard.edu/; http://plantcellbiology.masters.grkraj.org/;https://www.bio.sci.osaka-u.ac.jp
PDGF-b:
Signaling by the platelet-derived growth factor (PDGF) receptor; the unliganded receptor is monomeric and its tyrosine kinase catalytic activity is low (left). On binding to the receptor dimerizes; and its catalytic activity increases, and receptors Trans-phosphorylate each other on a number of different sites (specific), represented by pink circles (center). These phosphorylated sites (with one exception) serve to recruit cytosolic effector proteins (gray) that contain phosphotyrosine-specific modular binding domains (right). The exception is the activating phosphorylation, located on the catalytic domain of the receptor adjacent to the active site (red circle). Representative effectors depicted are: PI3K, regulatory subunit of phosphatidylinositol 3-kinase; GAP, Ras GAP, a GTPase-activating factor for Ras; PLC, phosphatidylinositol-specific phospholipase C-γ; Shp2, SH2-containing tyrosine phosphatase; Grb2, adaptor protein that recruits the Ras guanine-nucleotide exchange factor Sos. Src, Src-family non-receptor tyrosine kinases;
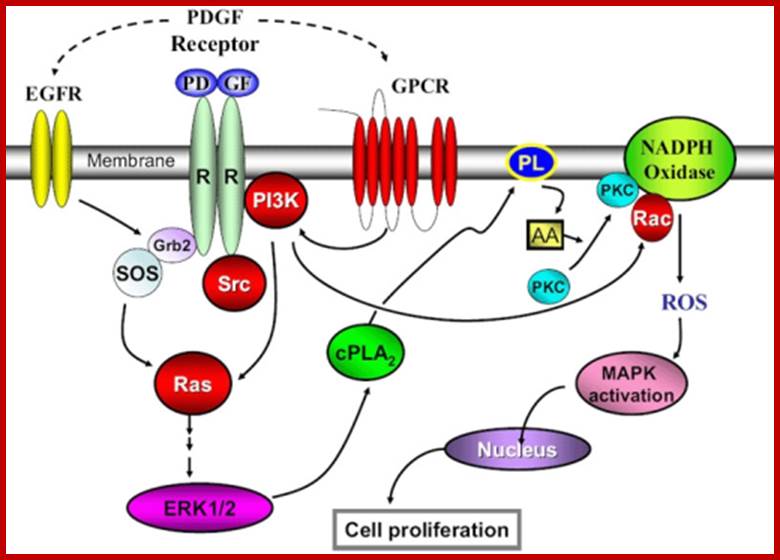
PDFG signaling pathway; http://www.sinobiological.com/
The four PDGF ligands-PDGFA-D are inactive in their monomeric forms. The PDGFs bind to the protein tyrosine kinase receptors PDGFRA and PDGFRB. These two receptor isoforms dimerize upon binding the PDGF dimer, leading to three possible receptor combinations, namely -AA, -BB and -AB. a critical tyrosine residue, thereby "unlocking" the kinase, leading to full enzymatic activity directed toward other tyrosine residues in the receptor molecules as well as other substrates for the kinase.
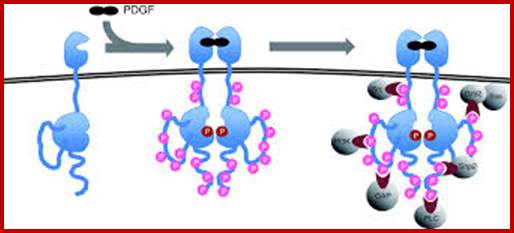
Dimerization can cause the activation of the kinase. Kinase activation is visualized as tyrosine phosphorylation of the receptor molecules, which occurs between the dimerized receptor molecules (trans phosphorylation). In conjunction with dimerization and kinase activation, the receptor molecules undergo conformational changes, which allow a basal kinase activity to phosphorylate http://jbiol.com/
When PDGF-b binds to its cell membrane receptor; it activates several components downstream, where phosphorylation by cellular domain of the receptor activates cytosolic s-mad proteins. Phosphorylated s-mad-2 interacts with phosphorylated s-mad-3 and produce dimers. The dimers in turn interact with unphosphorylated s-mad-4 factor and enter into the nucleus and activate several genes. PDFG receptor can also activated by Vascular Endothelial Factor (VGEF) acts using PDFG receptor.
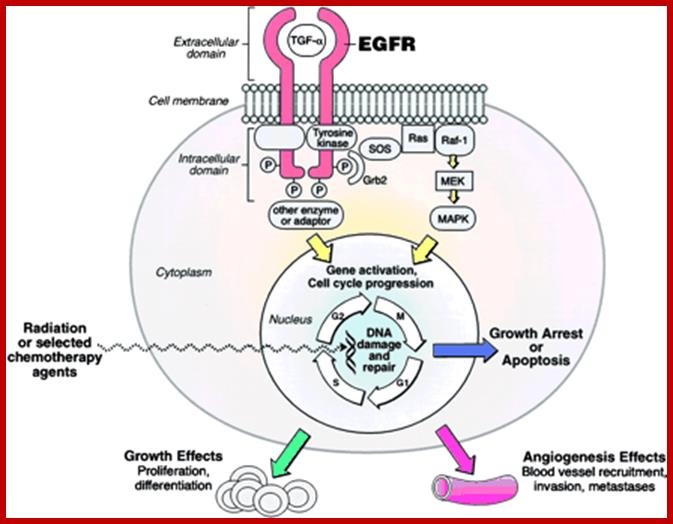
EGFR activation of cell division; http://www.cancertrials.ca/ffocus.htm
1. Activates the expression of expression of p15; it is very important for p15 proteins block transition from G1 to S, so block the progression of cell cycle events at the earliest stage itself.
2. They also activate genes whose products are inhibitors of plasminogen activator activated protease. When the plasminogen-activated protease is active, it digests most of the extra cellular matrix, thus give way for the cancer cells to move from one place to the other. But the expression of an inhibitor prevents the digestion of ECM (extra cellular matrix proteins), so cell adhesion and cell-to-cell contact is maintained, that prevents cancer cell migration and expansion.
3. The TGF-b activated s-mad transcriptional factors also activate gene expression whose product reinforce extra cellular matrix, so no cancer cells can squeeze through the extra cellular space in the tissue and prevent the spreading.
4. On the contrary, if the extra cellular signal is absent or lost none of the above said products are not produced and functions are not executed, thus cancer cells can easily migrate and spread through. It means this defect facilitates the spread of cancerous cells.
5. VEGF-A binds to PDFG receptors and activates PDGFG-AA to AB and BB receptors by activating tyrosine phosphorylation of PDFG-Rs.
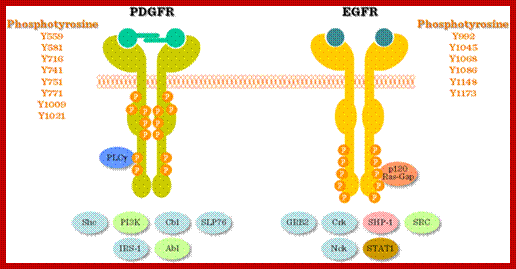
Growth factor receptors belong to the receptor tyrosine kinase family (for the details of growth factor receptor signalling -Receptor tyrosine kinases). http://www.tankonyvtar.hu/
Ligand binding leads to receptor dimerization, which induces phosphorylation of the cytosolic kinase domain and its activation (Figure above). Different receptors utilize different dimerization/activation strategies: for example PDGF is a dimer, which cross-links two cell surface PDGF receptor monomers; the binding of EGF to its receptor induces a conformational change, which promotes dimerization; FGF is complexed by heparin and cross links two FGF monomers; in case of insulin the receptor is already dimerized on the cell surface, ligand binding causes a conformational change and auto phosphorylation (for more details on insulin signalling see next chapter).
Wnt pathway:
Wingless gene (Wnt) from Drosophila is called by different names in different organisms. Wnt. gene product is a secreted signaling molecule. It binds to cell surface Frizzled receptors that activate disheveled (Dvl) proteins in human beings; actually these are activated by casein kinases (Cs 1 and 2). This inhibits the activity multiprotein complex (consisting of b-Catenin, Auxin, APC and glycogen synthase kinase (GSK-3b). Actually, this complex when in normal situations phosphorylates b-Catenin and thereby it is subjected ubiquitination and proteasome-mediated degradation. It is important to note that the b-Catenin is involved with C. Adherins in cell to cell adhesion.
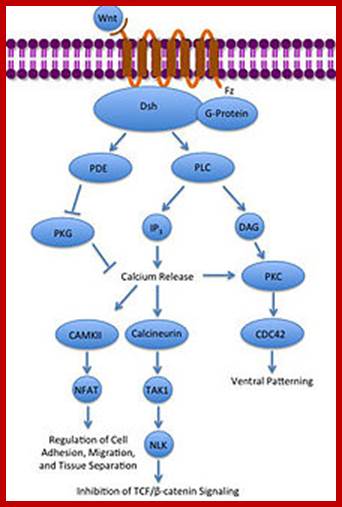
Noncanonical Wnt/calcium pathway; https://en.wikipedia.org
In the absence of Wnt signaling, the b-catenin accumulates in the cytoplasm and enters the nucleus and complexes with T-cell factors (TCF) and lymphoid enhancer factors (LEF) that results in the expression of cMyc and other genes required for continuous cell cycle events. The protein b-Catenin per se in association with other components is responsible for the activation of several genes, especially cMyc gene, responsible for inducing uninhibited cell cycle events.
The Wnt signaling pathways are critical in cell-cell signaling during normal development and embryogenesis and required for maintenance of adult tissue, therefore it is not difficult to understand why disruption in Wnt signaling pathways can promote human degenerative disease and cancer.
Signal Transducing Trans-membrane Receptor Proteins:
GPCR-Guanine protein coupled receptor, is seven-fold transmembrane proteins. The receptor is sensitive light, odors, pheromones, hormones and neurotransmitters. G proteins coupled receptors activate cAMP pathway and Phosphatidylinositol signal pathways. Research work on GPCR by Brain Kolbika and Robert Lefkowitz fetched Nobel prize in 2012. It is amazing that nearly 800 different human genes are predicted to code for GPCR. There are three subclasses, such as A-Rhodopsin like (largest),B-Secretin like and C- glutamate receptor like, others are Adhesion, Frizzled/Taste2 type and secretin like and few unclassified. Class A (or 1) (Rhodopsin-like), Class B (or 2) (Secretin receptor family), Class C (or 3) (Metabotropic glutamate/pheromone), Class D (or 4) (Fungal mating pheromone receptors), Class E (or 5) (Cyclic AMP receptors) and Class F (or 6) (Frizzled/Smoothened).
The protein has extracellular N- terminus followed by seven TM (7-TM) alpha helixes connected by three extracellular and three intracellular loops and its C- terminus (KKKRRK domain) is cytoplasmic end. It has cavity like structure to which ligands bind.

https://en.wikipedia.org
![]()
Signal transduction Pathway-GPCR; http://www.mun.ca/biology
There are more than 35-40 different ligands. They bind and activate this GPCR receptor. GPCR protein is bound to a trimer of alpha, beta and gamma subunits. It has Phosphatidylinositol and cAMP pathways. The alpha and gamma subunits are bound to PM- by lipid anchoring.
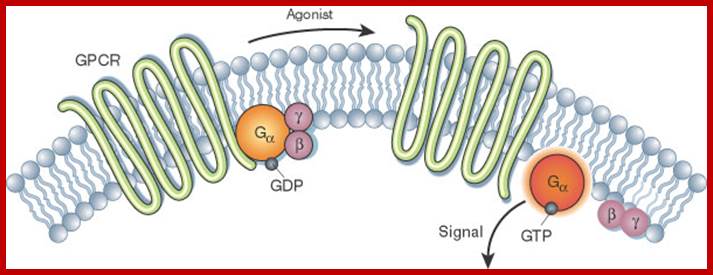
Activation of the G alpha subunit of a G-protein-coupled receptor
In unstimulated cells, the state of G alpha (orange circles) is
defined by its interaction with GDP, G beta-gamma (purple circles), and a
G-protein-coupled receptor (GPCR; light green loops). Upon receptor stimulation
by a ligand called an agonist, the state of the receptor changes. G alpha
dissociates from the receptor and G beta-gamma, and GTP is exchanged for the
bound GDP, which leads to G alpha activation. G alpha then goes on to activate
other molecules in the cell. © 2002 Nature
Publishing Group Li,
J. et al. The Molecule Pages database. Nature 420, 716-717 (2002). ![]()
G-protein’s alpha subunit binds to either GTP or GDP, when active it binds to GTP and when inactive it binds to GDP; in active form it is bound to GTP.
Activation of a single G protein activates the production of more than hundreds of second messenger molecules; the second messengers such a cAMP, Diacylglycerol (DAG) and Inositol (IP3), when activated initiate coordinated intercellular signaling pathways. One important, but common target is adenylyl cyclase a membrane associated enzyme. The cAMP has multiple functions on target substrate.
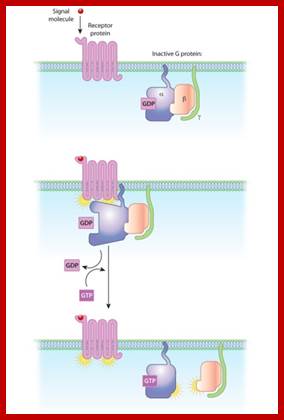
The relationships of G proteins to the plasma membrane:
In this diagram of G-protein-coupled receptor activation, the alpha, beta, and gamma subunits are shown with distinct relationships to the plasma membrane. After exchange of GDP with GTP on the alpha subunit, both the alpha subunit and the beta-gamma complex may interact with other molecules to promote signaling cascades. Note that both the alpha subunit and the beta-gamma complex remain tethered to the plasma membrane while they are activated. These activated subunits can act on ion channels in the cell membrane, as well as cellular enzymes and second messenger molecules that travel around the cell.
Receptor Kinases:
Many growth hormone receptors are transmembrane proteins with kinase activity at their cytosolic ends, but this need not be the case in all other membrane protein receptors. Epidermal growth factors, Platelet derived growth factors, Colony stimulating factors and many such factors act as mitogens. They bind to their membrane receptors and activate specific kinase activity.
EGF-Receptor Tyrosine Kinase:
Epidermal growth factor (EGF) receptor is a class-I receptor having N-terminal extracellular domain and a cytoplasmic domain with Tyrosine Kinase activity. This gene is also called c-erb-B. It has single trans-membrane domain. Ligand binding leads to dimerization and activation of Kinase activity at cytoplasmic side. This can lead to auto phosphorylation at it cytosolic domain. The cytosolic domains may contain protein-protein interacting domains such as SH2 and SH3. The active cytoplasmic domain transduces the signal to other proteins in the form of phosphorylation of target proteins at tyrosine moiety. There are 18-20 receptor kinases. The ligands are EGF, Insulin, PDGF, VEGF, FGF, HGF, Trk, Eph, AXL, LTK, TE, ROR, DDR, RET, KLG, RYK and MuSK.
But oncogenic proteins have deletions, either in the form of missing external domain or internal cytosolic domain or both, yet transmembrane domains remain as dimer and the protein remains active even without ligand binding.
Cell cycle specific Transcriptional regulation of Mitotic Genes:
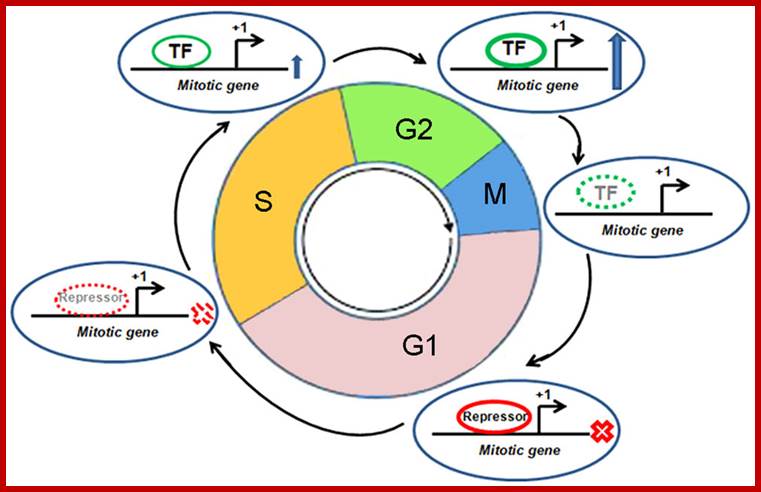
Cell cycle specific Transcriptional regulation of Mitotic Genes: cell cycle phases, Transcription start sites, Transcription Factors, Repressors are shown. X mark transcription off, Up arrow indicates transcription ‘ON’, dotted lines show reduction of recruitment of TFs or repressors, Proteins- contractile, functional enzymes, receptors, transport, defense, hormonal, enzymatic and structural htt;p://journal.frontiersin.org/
To decide to divide or not to divide: Quiescent state-non dividing state called Go, the decision not to divide is due to not receiving required growth factors stimuli or signals. What happens in cells that "decide" to divide? If the decision is to divide (e.g., if a growth factor signal reaches cell), then the cell will go into S phase, and DNA synthesis will begin. Once this happens, the cell must go through the cell division process, all the way through to the end. However, each stage of the cell cycle must be successfully completed, before the next stage can be entered, and each step is carefully regulated. The cell has various mechanisms, called check points, that allow it to proceed from one stage to the next only after the previous one has been confirmed to be completed. http://eishinoguchi.com
Checkpoint Controls:
-1st:
Between G1 & S
---Looks for favorable environment
-2nd: Between
G2 & M
---Looks for favorable environment
---Looks to see if all DNA has been replicated &
replicated correctly
-3rd: In
the middle of M
---Metaphase to anaphase transition
--Looks to see if all chromosomes have been attached to
spindles properly
What does the cell cycle checkpoint control system depend on?
1. Cyclically
activated cyclin-dependent kinases (Cdks)
2. Cyclical proteolytic events
3. Transcriptional regulation
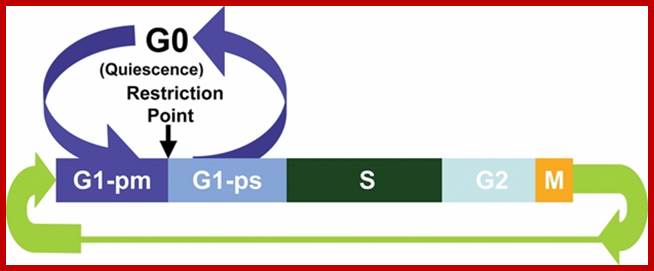
Time period-G1 -9hrs, S phase 10hrs and G24 and 1 half, M phase- one-half hrs; http://www.ncbi.nlm.nih.gov/
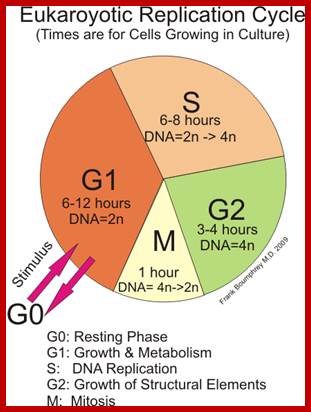
http://study.com/academy
Mammalian cell cycle: The mammalian cell cycle originally described as consisting of 2 parts: mitosis and interphase. During interphase, the genome is duplicated and was called S phase for synthesis. G1 and G2 were proposed as the gaps between mitosis and S phase and between S phase and mitosis, respectively. G1 is usually where critical decisions are made as to whether to enter a resting quiescent stage known as G0 or to continue cycling and commit to replicating the genome and mitosis. The point in G1 where this growth factor–dependent decision is made is known as the restriction point (R). G1 has been described as consisting of 2 parts on either side of R, where the first part of G1 is known as G1-pm for post mitotic, and the second part is known as G1-ps for pre-S.
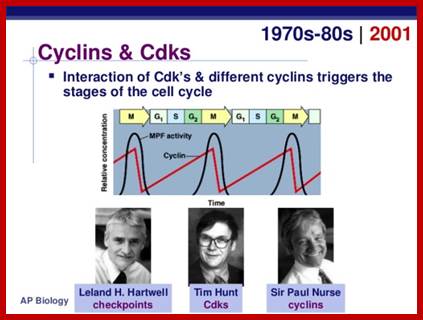
Great scietists explained diffent cyclins bind to specific CDKs and activate each of the steps in cell cycle. Two key components of the cell-cycle control system; Simplified view of the core of the cell-cycle control system; http://image.slidesharecdn.com/
A complex of cyclin with Cdk acts as a protein kinase to trigger specific cell-cycle events. Without cyclin, Cdk is inactive.
Cdk associates successively with different cyclins to trigger the different events of the cycle. Cdk activity is usually terminated by cyclin degradation. For simplicity, only the cyclins that act in S phase (S-cyclin) and M phase (M-cyclin) are shown, and they interact with a single Cdk; as indicated, the resulting cyclin-Cdk complexes are referred.
A key group of proteins in this regulatory process are Cyclin-dependent kinase (CDK) proteins. Although CDK levels remain consistent throughout the cell cycle, Cyclin levels differ based on the stage of the cell cycle and though this rising and falling of these levels they activate or deactivate CDK’s. Different CDK and different Cyclins are needed for each phase of the cell cycle. CDK4 and CDK6 both bind to Cyclin D1, D2, and D3 in the G1 phase and are needed for entry into the G1 phase. Differing from other cyclins which are expressed periodically in the cell cycle, if growth factor simulation is persistent then cyclin D will continue to be synthesized. CDK2 and its activating protein Cyclin E are essential for the progression from G1 phase to the S phase. CDK2 and cyclin A are important in the S phase. As for the transition from the G2 to Mitosis CDK1 and its activating protein Cyclin A are required and mitosis is further regulated by the CDK1 and Cyclin B complex.
Information about the completion of cell-cycle events, as well as signals from the environment, can cause the control system to arrest the cycle at specific checkpoints. The most prominent checkpoints occur at locations marked with yellow boxes.
Checkpoints in the cell-cycle control system;
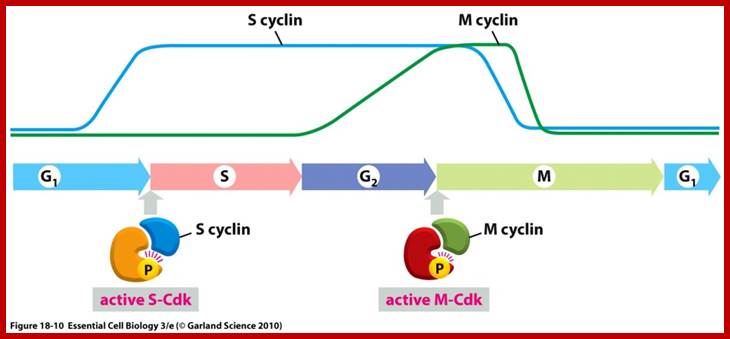
Role of different cyclins and CDKs at different phases of Cell-Cycle; http://oregonstate.edu/
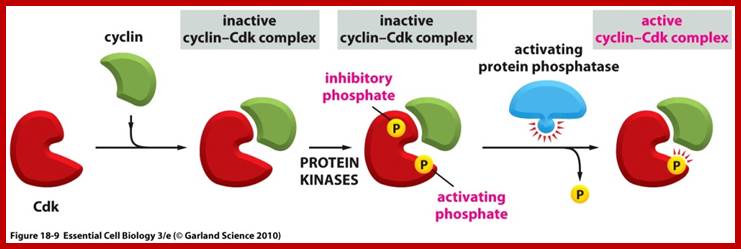
Activation of CDKs by cyclins; http://oregonstate.edu/
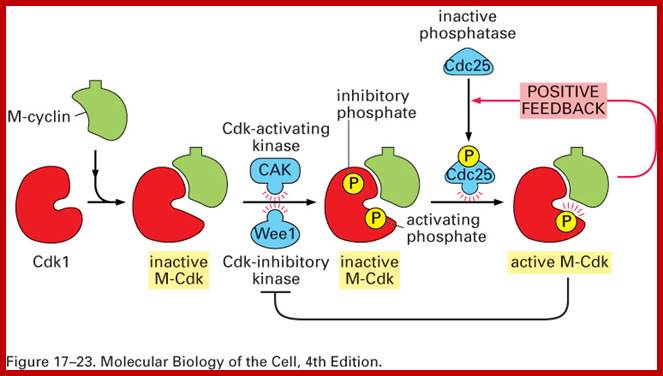
Factors that act in activating CDK-Cyclin dimers; http://www.pha.jhu.edu/~ghzheng

The major events of the cell cycle: The major events of the cell cycle are regulated by successive waves of kinase and ubiquitin ligase activity. G1-cyclin–CDK activity is required to initiate the cell cycle and activate B-type-cyclin–CDK activity. Low levels of B-type-cyclin–CDK activity are sufficient to trigger S phase, but tyrosine phosphorylation by Wee1 prevents full activation, preventing premature mitosis. Full CDK activation triggers mitosis and activates APC, which triggers anaphase and feeds back to inactivate CDK activity. Inactivation of CDK allows exit from mitosis and the reestablishment of interphase chromosome and nuclear structure in G1 phase. See Box 1 for description of the stages of mitosis; http://www.ncbi.nlm.nih.gov/ http://greatcourse.cnu.edu.cn/
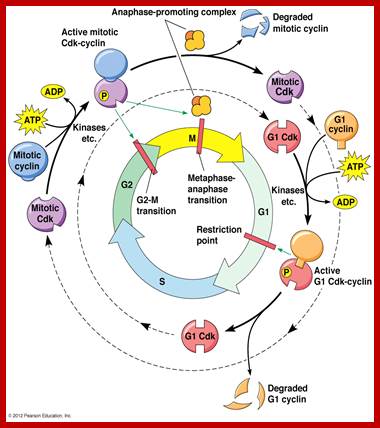
Role of different Cyclin with different CDKs in cell cycle process; http://www.mun.ca/biology http://www.mun.ca/biology

Conditions or factors that leads to check the cell cycle progress at specific stages; http://oregonstate.edu/
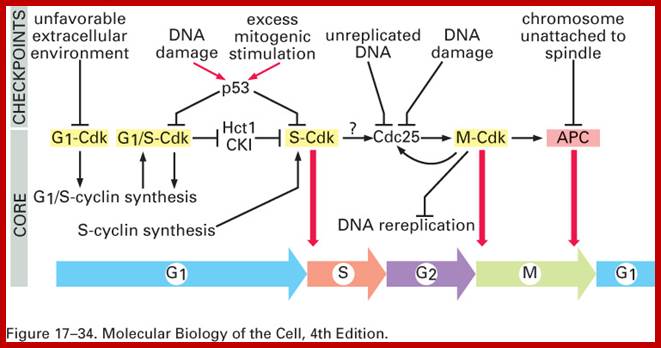
Check points internal- at G1, G2/M and spindle; G1 -9hrs, S phase 10hrs and G2,4 and 1half, M phase- one-half hrs,Check point proteins and cyclins/CDKs at specific phases or stages of cell cycle; http://www.pha.jhu.edu/~ghzheng; http://biowiki.ucdavis.edu/
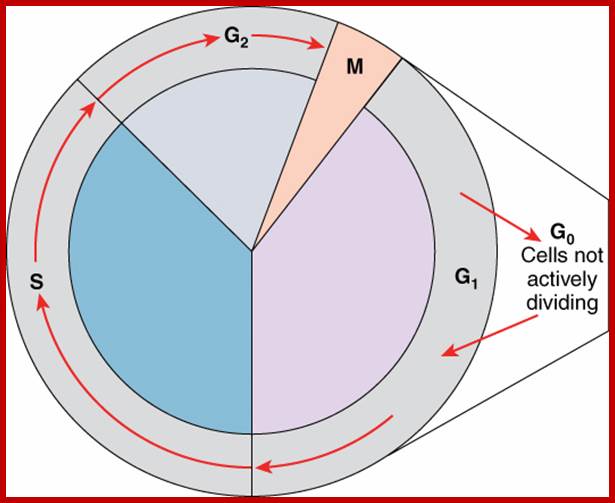
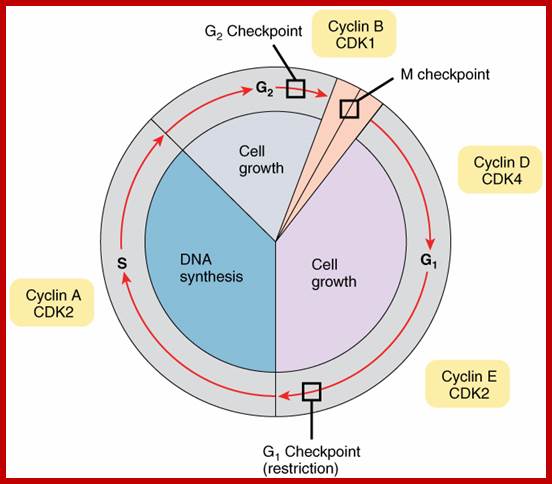
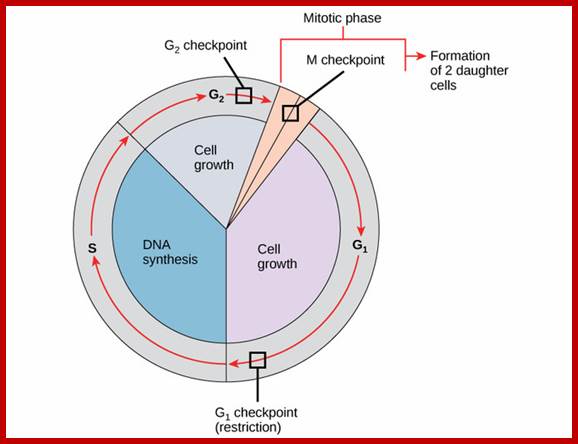
“Regulation of the Cell Cycle at Internal Che
Checkpoints; The cell-cycle is controlled at three checkpoints. The integrity
of the DNA is assessed at the G1 checkpoint. Proper chromosome duplication is
assessed at the G2 checkpoint. Attachment of each kinetochore to a spindle
fiber is assessed at the M checkpoint”.
https://www.boundless.com/-
Retrieved 09 Oct. 2016
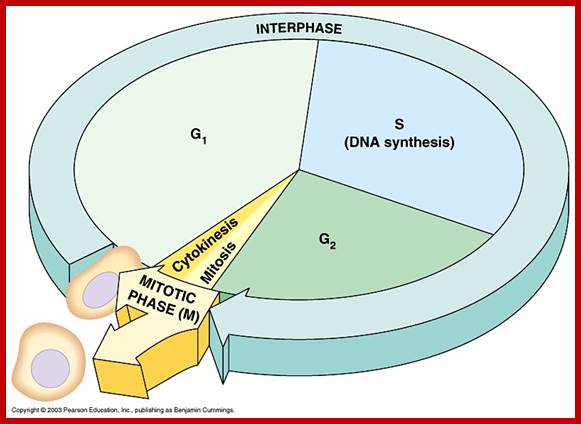
http://cursa.ihmc.us/; https://www.spandidos-publications.com/

http://image.slidesharecdn.com/
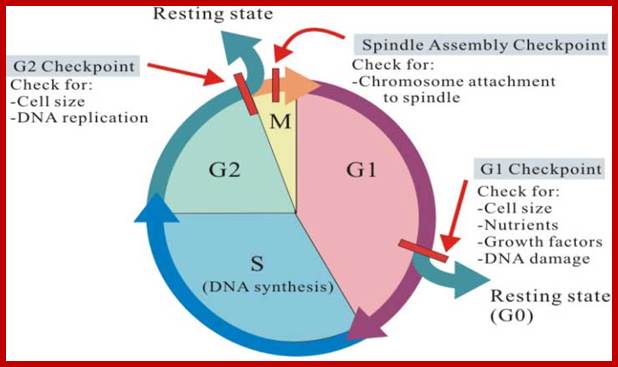
Cell Division may be regulated by internal and external factors. http://gleesonbiology.pbworks.com/w
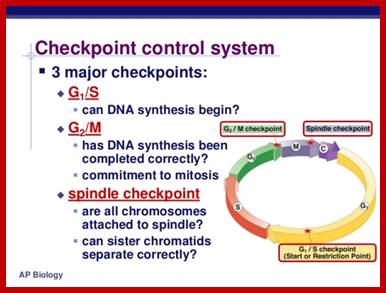
http://image.slidesharecdn.com/

http://image.slidesharecdn.com/

http://image.slidesharecdn.com/
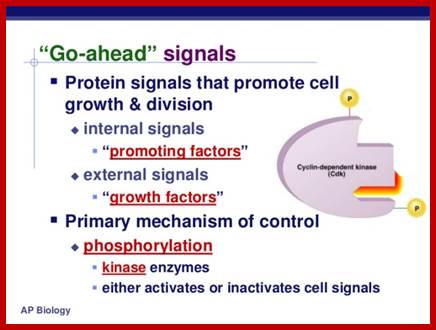
http://image.slidesharecdn.com/
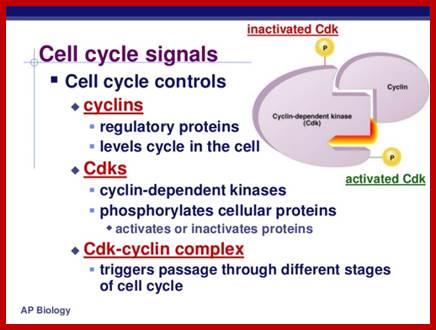
http://image.slidesharecdn.com/
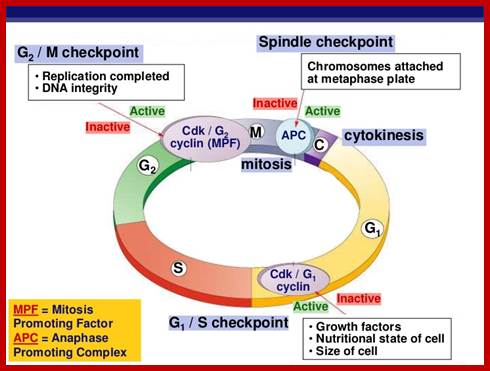
http://image.slidesharecdn.com/
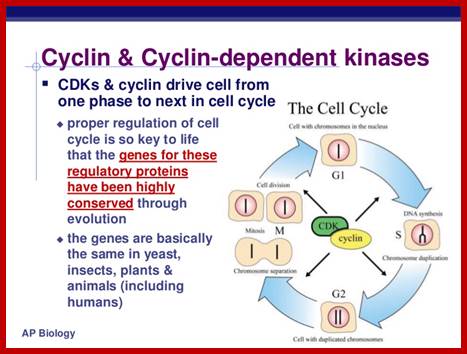
http://image.slidesharecdn.com/
Irradiation induces G1 and G2 cell cycle checkpoint activation and DNA repair. Most cancer cells are defective in G1 checkpoint, commonly due to the mutations/alterations of the key regulators of the G1 checkpoint (in blue), but contain a functional G2 checkpoint. https://www.researchgate.net; https://www.spandidos-publications.com

http://www.slideshare.net/ NeoplasisII
Irradiation induces G1 and G2 cell cycle checkpoint activation and DNA repair. Most cancer cells are defective in G1 checkpoint, commonly due to the mutations/alterations of the key regulators of the G1 checkpoint (in blue), but contain a functional G2 checkpoint. https://www.researchgate.net
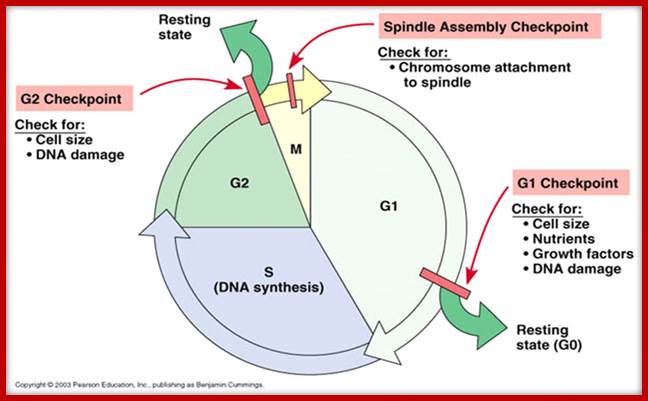
http://www.sci.nu.ac.th/ https://oncogenesandcancer.files.wordpress.com
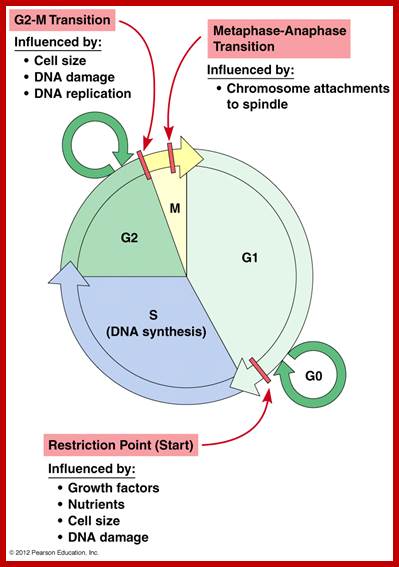
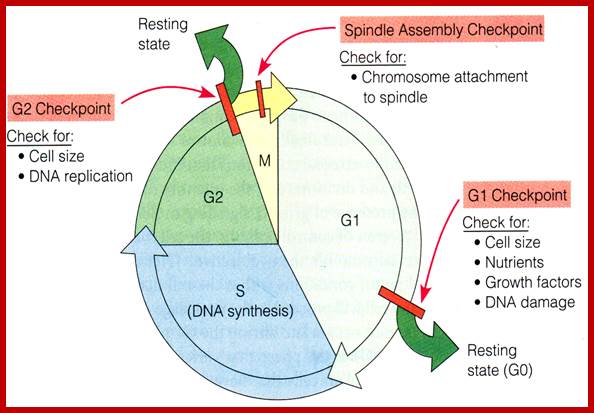
Oncogenes and cancer.wordpressd.com
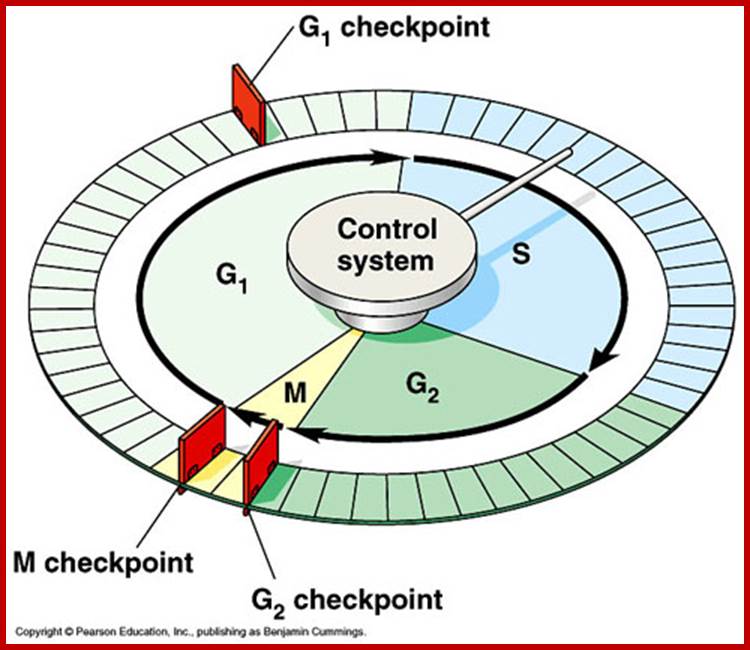
http://abenagh.pbworks.com/
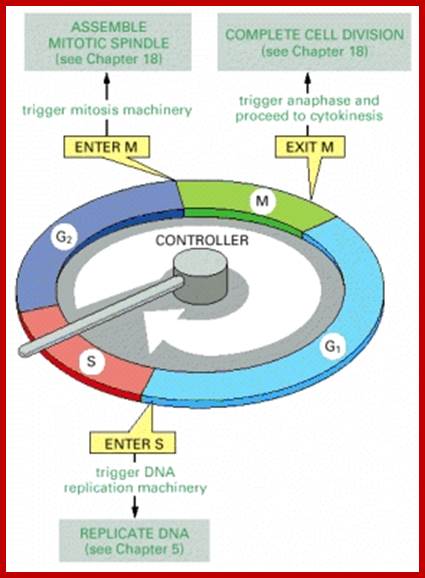
The essential processes of the cell cycle—such as DNA replication, mitosis, and cytokinesis—are triggered by a cell-cycle control system. By analogy with a washing machine, the cell-cycle control system is shown here as a central arm—the controller—that rotates clockwise, triggering essential processes when it reaches specific points on the outer dial. http://www.ncbi.nlm.nih.gov/
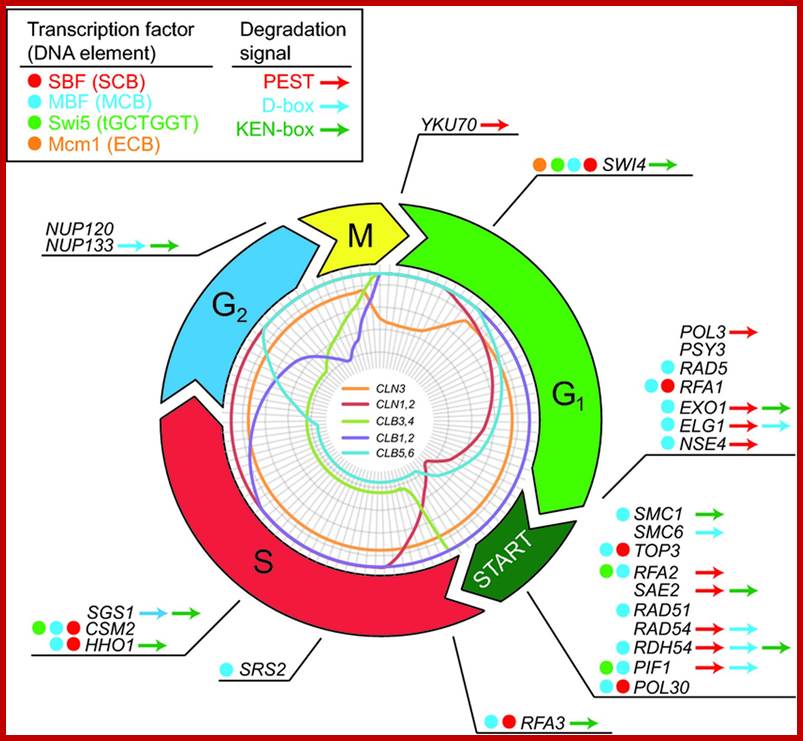
Transcriptional and proteolytic cell cycle regulation of HR. The transcriptional regulation of HR during the cell cycle in budding yeast. The inner circle illustrates the transcriptional levels of the cyclins Cln1-3 and Clb1-6 through the cell cycle and is adapted from (Richardson et al., 1992; Schwob & Nasmyth, 1993; Tyers et al., 1993; McInerny et al., 1997; Fitch et al., 2003). The cell-cycle-regulated genes involved in HR are derived from genome-wide microarray data set performed by Cho et al., 1998; Spellman et al.,1998; Pramila et al., 2002; and de Lichtenberg et al., 2003. Four transcription factors and corresponding upstream promoter elements are shown: SBF, MBF, Swi5, and Mcm1 (Spellman et al., 1998). A colored dot in front of each gene name indicates whether the gene expression is controlled by one of the four transcription factors. Protein degradation signals of the D-box, KEN-box or PEST region types are illustrated by the presence of a colored arrow following each gene. The annotation of degradation signals in known proteins related to HR was based on the consensus sequences: D-box, RXXLXXXXN; KEN-box, KENXXXN; and PEST region, rich in proline, glutamic acid, serine, and threonine (Jensenet al., 2006); http://femsre.oxfordjournals.org/
External- growth promoting hormones leads to increase in cell size, and overcrowding of cells. G1- Cell size-DNA damage and repair, G2 – enter into M-phase- DNA damage repaired or not, M- check point- at metaphase- also called spindle check point- correct attachment of spindle fibers to kinetochores.
Controlled at three checkpoint; The integrity of the DNA is assessed at the G1 checkpoint. Proper chromosome duplication is assessed at the G2 checkpoint. Attachment of each kinetochore to a spindle fiber is assessed at the M checkpoint. http://biowiki.ucdavis.edu/ Among many proteins, important ones are RB, p53 and CHEK proteins.
G1 Check point proteins: During early G1, the transcriptional repressors Rb (retinoblastoma), p107 and p130, known as pocket proteins, bind to the E2F transcription factors to prevent G1-to-S transition. Rb binds and represses activator E2F transcription factors (E2F1-3), while p107 and p130 bind E2F4 and E2F5 to form complexes which repress transcription of G1-to-S promoting factors.
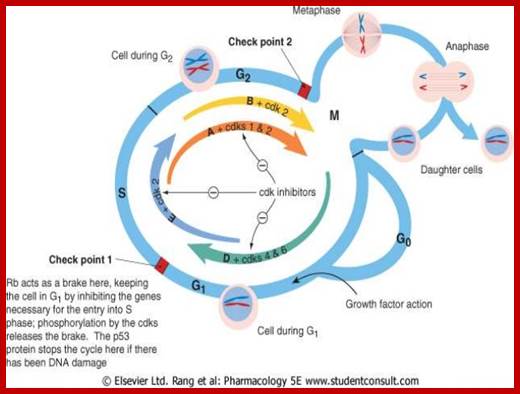
http://image.slidesharecdn.com/
When DNA damage occurs, or when the cell detects any defects which necessitate it to delay or halt the cell cycle in G1, arrest occurs through several mechanisms. The rapid response involves phosphorylation events that initiate with either kinase ATM (Ataxia telangiectasia mutated) or ATR (Ataxia Telangiectasia and Rad3 related), which act as sensors, depending on the type of damage.
These kinases phosphorylate and activate the effector kinases Chk2 and Chk1, respectively, which in turn phosphorylate the phosphatase Cdc25A, thus marking it for ubiquitination and degradation.
Rad1 is a component of the 9-1-1 cell-cycle checkpoint response complex, which plays a role in checkpoint activation that permits DNA-repair pathways to prevent cell cycle progression in response to DNA damage and replication stress. The 9-1-1 complex is recruited to DNA lesions upon damage by the Rad17 (Rad24 in budding yeast)-replication factor C (RFC) clamp loader complex. The 9-1-1 complex is necessary for the recruitment of C12orf32/RHINO to sites of double-stranded breaks (DSB) occurring during the S phase. Rad1 isoform 1 possesses 3'->5' double stranded DNA exonuclease activity; https://www.ebi.ac.uk
G2 check point Prots;
In most species, G2, in which cells increase protein synthesis and undergo rapid growth in preparation for mitosis, at this point of cell division check point G2/M operates. The G2/M checkpoint, also known as the DNA damage checkpoint, ensures that the cell underwent all of the necessary changes during the S and G2 phases and is ready to divide.
Metaphase check point: The mitotic spindle checkpoint occurs at the point in metaphase where all the chromosomes should/have aligned at the mitotic plate and be under bipolar tension. The tension created by this bipolar attachment is what is sensed, which initiates the anaphase entry. To do this, the sensing mechanism ensures that the anaphase-promoting complex (APC/C) is no longer inhibited, which is now free to degrade cyclin B, which harbors a D-box (destruction box), and to break down Securin. The latter is a protein whose function is to inhibit separase, which in turn cuts the Cohesins, the protein composite responsible for cohesion of sister chromatids. Once this inhibitory protein is degraded via ubiquitination and subsequent proteolysis; then separase acts and separates sister chromatids. After the cell has split into its two daughter cells, the cell enters G1.
The mechanisms by which mitotic entry is prevented in response to DNA damage are similar to those in the G1/S checkpoint. DNA damage triggers the activation of the aforementioned ATM/ATR pathway, in which ATM/ATR phosphorylate and activate the Chk1/Chk2 checkpoint kinases. Chk1/2 phosphorylates cdc25 which, in addition to being inhibited, is also sequestered in the cytoplasm by the 14-3-3 proteins. 14-3-3 are upregulated by p53, which, as previously mentioned, is activated by Chk1 and ATM/ATR. p53 also transactivates p21, and both p21 and the 14-3-3 in turn inhibit cyclin B-cdc2 complexes through the phosphorylation and cytoplasmic sequestering of cdc2. In addition, the inactivation of cdc25 results in its inability to dephosphorylate and activate cdc2. Finally, another mechanism of damage response is through the negative regulation of Plk1 by ATM/ATR, which in turn results in the stabilization of Wee1 and Myt1, which can then phosphorylate and inhibit cdc2, thus keeping the cell arrested in G2 until the damage is fixed; https://en.wikipedia.org
Cells carefully monitor the alignment of chromosomes on the mitotic spindle to ensure that their chromosomes are properly segregated at anaphase. Detachment of kinetochores from microtubules or depolymerization of the mitotic spindle activates a checkpoint mechanism that arrests the cell cycle prior to the initiation of anaphase. This ‘spindle assembly checkpoint’ allows the cell time to correct the offending lesion so that it can proceed through mitosis without chromosome mis-segregation. Proteins involved in the spindle assembly checkpoint have been isolated in budding yeast and in vertebrates. Cells lacking components of the spindle assembly checkpoint can proceed through mitosis in the absence of a spindle or in the presence of unattached chromosomes.
Recent work has shown that two of the spindle assembly checkpoint proteins, Mad2 and Bub1, localize to the kinetochores of vertebrate cells for part of mitosis. Prophase and prometaphase chromosomes show kinetochore localization of Mad2 and Bub1, but as chromosomes align on the metaphase plate the kinetochore staining disappears and does not reappear until the next cell cycle. Misaligned chromosomes show Mad2 and Bub1 localization at the kinetochore even when all other chromosomes reside on the metaphase plate. These results suggest that the kinetochore both monitors the attachment of the chromosome to the spindle and signals the cell to arrest until chromosomes are properly aligned.
Mad2 and Bub1 proteins reside at kinetochores and mediate checkpoint arrest of the cell cycle. (a) During a normal mitosis, Mad2 and Bub1 localize to kinetochores in prophase and prometaphase. Once all chromosomes are attached to the spindle, cells proceed through anaphase. (b) Misaligned chromosomes signal spindle assembly checkpoint arrest. Chromosomes that are not properly attached to the spindle maintain Mad2 at the kinetochore and the cell arrests in mitosis. (c) Spindle depolymerization causes mitotic arrest. In the absence of a spindle, Mad2 and Bub1 remain at the kinetochores of chromosomes and the cell arrests in mitosis. The checkpoint arrest can be overcome by two mechanisms: by error correction, the cell can correct the offending lesion and process through a normal mitosis; by adaptation, the cell can override the checkpoint arrest, in the presence of the defect, resulting in an abnormal mitosis with the potential for chromosome loss. http://www.cell.com/
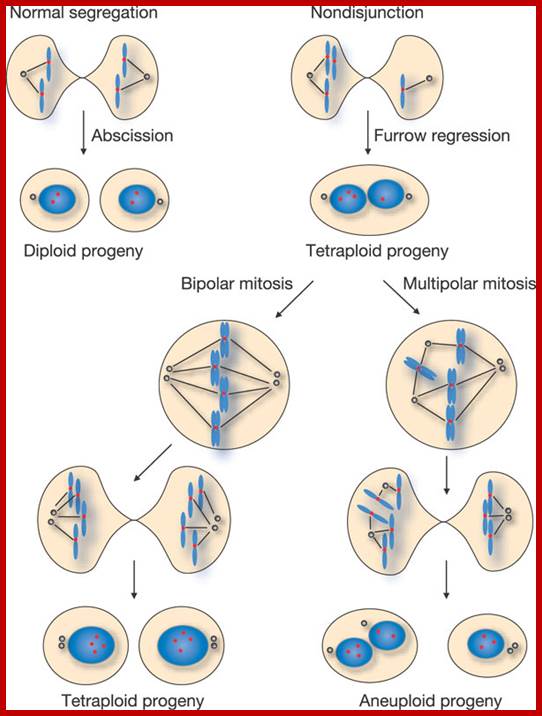
Mechanisms by which tumor suppressor mutation-associated microtubule defects could contribute to tumor capabilities. Symbols and associated tumor cell capabilities are adapted from the Hanahan and Weinberg hallmarks wheel (with permission from Elsevier) (Hanahan and Weinberg, 2000), with the addition of a separate symbol for genomic instability. Where applicable, the roles microtubules play in preventing development of these capabilities are listed. Qinghua Shi & Randall W. King http://www.nature.com/

Mitotic defects leading to whole-chromosome aneuploidy. (A) An anaphase cell that has undergone proper chromosome segregation and begun anaphase. (B–D) Tumor suppressor-associated mitotic spindle defects, (B) Individual chromosomes can segregate incorrectly owing to failed attachments of kinetochore microtubules. (C) Multiple chromosomes can segregate to the wrong daughter cell owing to tripolar spindle formation. (D) Cytokinesis can fail, resulting in a single, tetraploid daughter cell. http://dmm.biologists.org/
Mad2 and Bub1 proteins reside at kinetochores and mediate checkpoint arrest of the cell cycle. (a) During a
normal mitosis, Mad2 and Bub1 localize to kinetochores in prophase and prometaphase. Once all chromosomes are attached to the spindle, cells proceed through anaphase. (b) Misaligned chromosomes signal spindle assembly checkpoint arrest. Chromosomes that are not properly attached to the spindle maintain Mad2 at the kinetochore and the cell arrests in mitosis. (c) Spindle depolymerization causes mitotic arrest. In the absence of a spindle, Mad2 and Bub1 remain at the kinetochores of chromosomes and the cell arrests in mitosis. The checkpoint arrest can be overcome by two mechanisms: by error correction, the cell can correct the offending lesion and process through a normal mitosis; by adaptation, the cell can override the checkpoint arrest, in the presence of the defect, resulting in an abnormal mitosis with the potential for chromosome loss. http://www.cell.com/current-biology
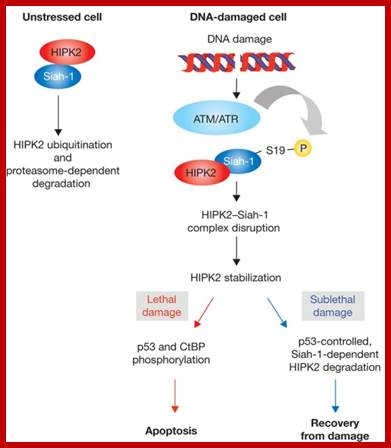
Control of HIPK2 stability by ubiquitin ligase Siah-1 and checkpoint kinases
ATM and ATR; www.nature.com
![]()
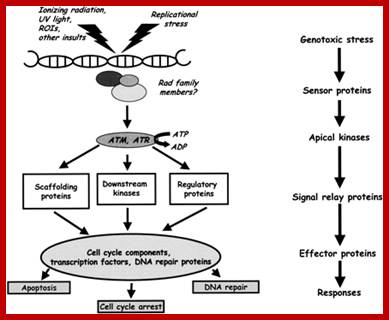
P53 gets activated due to the said factors as shown in the figure, and when p53 is activated, one finds what it does. Factors that act on p53 and activate p53; http://www.biotechniques.com/ Cell cycle checkpoint signaling through the ATM and ATR kinases; cell cycle check point signaling pathway- Reactive Oxygen; http://genesdev.cshlp.org/
Cell cycle checkpoint signaling through the ATM and ATR kinases; cell cycle check point signaling pathway- Reactive Oxygen:
As mentioned earlier, ATM and ATR belong to a structurally unique family of protein serine–threonine kinases whose catalytic domains share a clear evolutionary relationship with those of mammalian and yeast phosphoinositide 3-kinases.
In contrast to ATR, ATM seems dedicated to providing the cell with a rapid protective response to an extremely lethal form of DNA damage, the DNA dsb. Although ATM research has focused heavily on the response to IR-induced DNA dsbs, it is likely that, outside of the laboratory setting, the intrinsic accoutrements of life as a multicellular organism, such as continuous assaults by oxygen-derived radicals, and the need to generate a diverse repertoire of antigen-responsive lymphocytes, generated evolutionary pressure for the development of a checkpoint signaling module geared toward sounding the DNA dsb alarm at the first sign of trouble. The identification of ATM as a key regulator of p53 in cells that have incurred DNA dsbs not only links ATM to the G1 checkpoint, but also suggests that ATM participates directly in making life and death decisions in certain cell types, particularly thymocytes and early-stage neurons. In A-T patients, disruption of the p53-dependent pathways that would normally direct irreparably damaged cells to undergo apoptosis during embryonic or early postnatal life may sow the seeds for the neurodegeneration and cancer predisposition that are so characteristic of this disease. An intriguing possibility, which demands additional attention, is that ATM plays a more general role in cyto-protection against oxidative stress, possibly by inducing the expression of NFκB-regulated genes.
In eukaryotic cells, maintenance of genomic stability relies on the coordinated action of a network of cellular processes, including DNA replication, DNA repair, cell-cycle progression, and others. The DNA damage response (DDR) signaling pathway orchestrated by the ATM and ATR kinases is the central regulator of this network in response to DNA damage. Both ATM and ATR are activated by DNA damage and DNA replication stress, but their DNA-damage specificities are distinct and their functions are not redundant. Furthermore, ATM and ATR often work together to signal DNA damage and regulate downstream processes. Here, we will discuss the recent findings and current models of how ATM and ATR sense DNA damage, how they are activated by DNA damage, and how they function in concert to regulate the DDR. https://www.ncbi.nlm.nih.gov.
Highly selective small molecule inhibitors of ATM and ATR are currently in preclinical and clinical development, respectively. Preclinical data have provided a strong rationale for clinical testing of these compounds both in combination with radio- or chemotherapy, and in synthetic lethal approaches to treat tumour with deficiencies in certain DDR components. Whole genome sequencing studies have reported that mutations in DDR genes occur with a high frequency in many common tumour types, suggesting that a synthetic lethal approach with ATM or ATR inhibitors could have widespread utility, providing that appropriate biomarkers are developed.
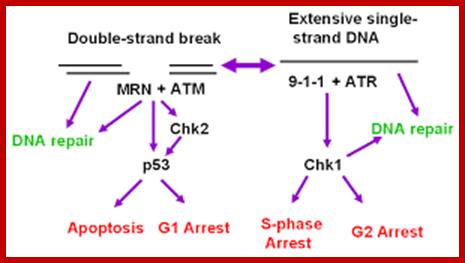
Use of ATR inhibitors in treating cancer patients; Jill M. Wagner and Scott H. Kaufmann www.mdpi.com
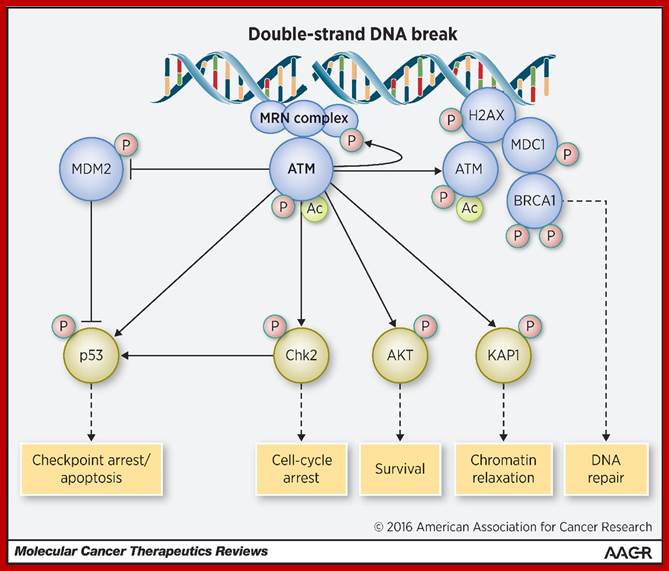
Once activated, ATM phosphorylates many downstream effectors, which in turn phosphorylate their own targets. The PIKK domain of ATM recognizes serine-glutamine (SQ) and threonine-glutamine (TQ) motifs of many proteins, including ones involved in cell-cycle checkpoint arrest (e.g., Chk1 and Chk2), DNA repair (BRCA1 and RAD51), and apoptosis (p53; ref.15). It also has been determined that there are in fact over 700 targets phosphorylated following DSBs, and that ATM modulates networks not immediately involved in DNA repair like the insulin-like growth factor or other metabolic and stress-response pathways (16). The plethora of ATM targets is likely a means of coordinating multiple pathways at times of DNA repair or genomic stress. There are also redundancies and collaboration between ATM and other members of the PIKK family, including catalytic subunit of the DNA-dependent protein kinase (DNA-PKc) and ATM-related (ATR), which are activated in responses to other sources of genotoxic stress. These redundancies may represent therapeutic targets for treating cancers that have lost ATM function.; http://mct.aacrjournals.org/
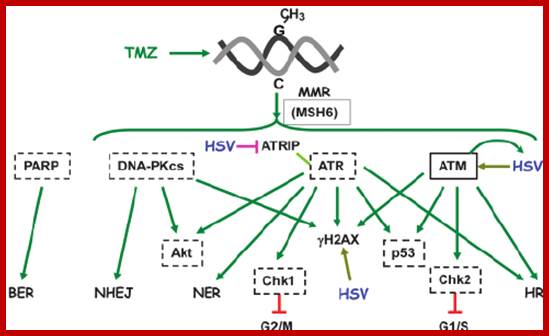
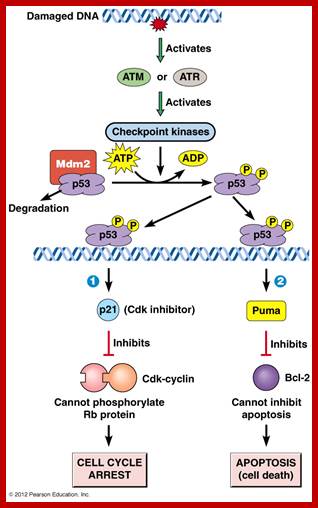
Role of ATM/ATR cell cycle fate; http://www.annualreviews.org/
DNA repair mechanisms include direct repair, base excision repair, nucleotide excision repair, double-strand break repair, and cross-link repair. The DNA damage checkpoints employ damage sensor proteins, such as ATM, ATR, the Rad17-RFC complex, and the 9-1-1 complex, to detect DNA damage and to initiate signal transduction cascades that employ Chk1 and Chk2 Ser/Thr kinases and Cdc25 phosphatases. The signal transducers activate p53 and inactivate cyclin-dependent kinases to inhibit cell cycle progression from G1 to S (the G1/S checkpoint), DNA replication (the intra-S checkpoint), or G2 to mitosis (the G2/M checkpoint). In this review the molecular mechanisms of DNA repair and the DNA damage checkpoints in mammalian cells are analyzed.
Internal Check points: Internal check points-DNA damage repair-G1 check point, G2 check point chromosome replication, M-check point sister chromatids are properly attached to MTs.
The cell cycle proceeds by a defined sequence of events where late events depend upon completion of early events. The aim of the dependency of events is to distribute complete and accurate replicas of the genome to daughter cells. To monitor this dependency, cells are equipped with the checkpoints that are set at various stages of the cell cycle. When cells have DNA damages that have to be repaired, cells activate DNA damage checkpoint that arrests cell cycle. According to the cell cycle stages, DNA damage checkpoints are classified into at least 3 checkpoints: G1/S (G1) checkpoint, intra-S phase checkpoint, and G2/M checkpoint. Upon perturbation of DNA replication by drugs that interfere with DNA synthesis, DNA lesions, or obstacles on DNA, cells activate DNA replication checkpoint that arrests cell cycle at G2/M transition until DNA replication is complete. There are more checkpoints such as Spindle checkpoint and Morphogenesis checkpoint. The spindle checkpoint arrests cell cycle at M phase until all chromosomes are aligned on spindle. This checkpoint is very important for equal distribution of chromosomes. Morphogenesis checkpoint detects abnormality in cytoskeleton and arrests cell cycle at G2/M transition. http://eishinoguchi.com/checkpoint.htm
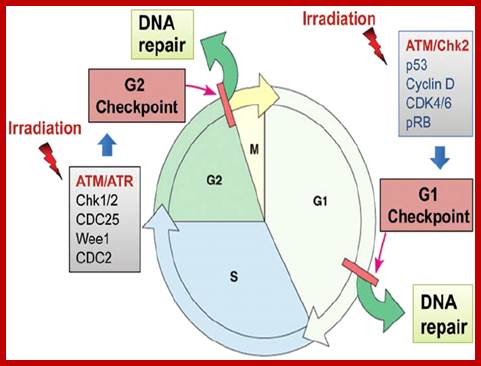
Irradiation induces G1 and G2 cell cycle checkpoint activation and DNA repair. Most cancer cells are defective in G1 checkpoint, commonly due to the mutations/alterations of the key regulators of the G1 checkpoint (in blue), but contain a functional G2 checkpoint. https://www.researchgate.net; https://www.spandidos-publications.com
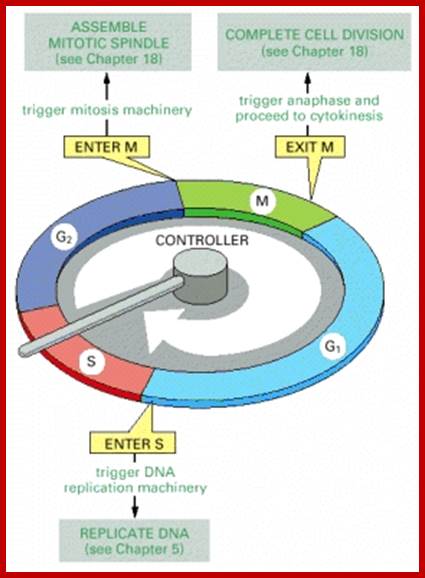
The essential processes of the cell cycle—such as DNA replication, mitosis, and cytokinesis—are triggered by a cell-cycle control system. By analogy with a washing machine, the cell-cycle control system is shown here as a central arm—the controller—that rotates clockwise, triggering essential processes when it reaches specific points on the outer dial. http://www.ncbi.nlm.nih.gov/
Information about the completion of cell-cycle events, as well as signals from the environment, can cause the control system to arrest the cycle at specific checkpoints. The most prominent checkpoints occur at locations marked with yellow boxes. Checkpoints in the cell-cycle control system
DNA Damage checkpoint;
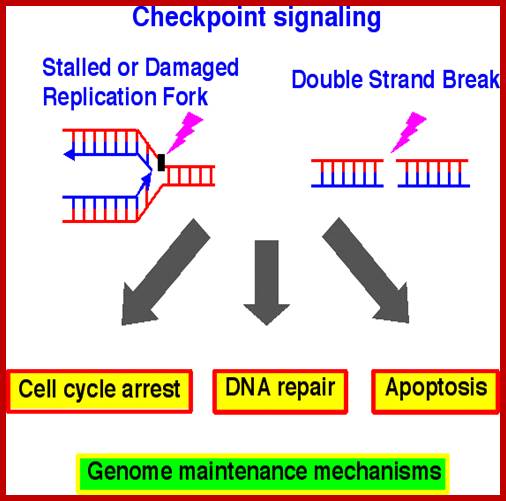
DNA replication and chromosome distribution are indispensable events in the cell cycle control. Cells must accurately copy their chromosomes, and through the process of mitosis, segregate them to daughter cells. The checkpoints are surveillance mechanism and quality control of the genome to maintain genomic integrity. Checkpoint failure often causes mutations and genomic arrangements resulting in genetic instability. Genetic instability is a major factor of birth defects and in the development of many diseases, most notably cancer. Therefore, checkpoint studies are very important for understanding mechanisms of genome maintenance as they have direct impact on the ontogeny of birth defects and the cancer biology. http://www.eishinoguchi.com/
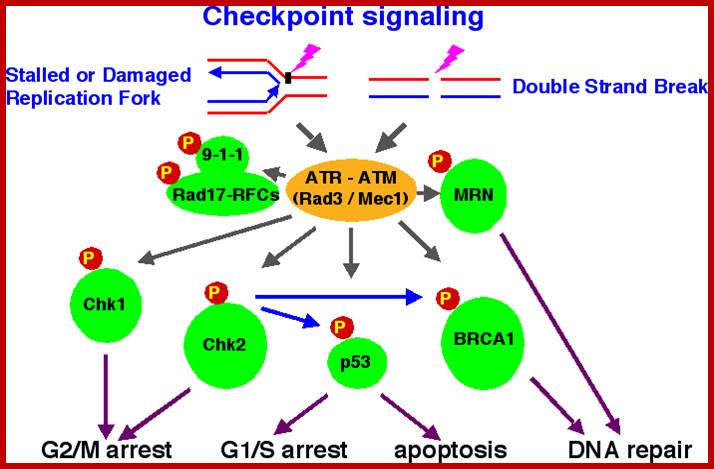
Accurate duplication of eukaryotic genome is a challenging task, given that environment of cell growth and division is rarely ideal. Cells are constantly under the stress of intrinsic and extrinsic agents that cause DNA damage or interference with DNA replication. To cope with these assaults, cells are equipped with DNA maintenance checkpoints to arrest cell cycle and facilitate DNA repair pathways. DNA maintenance checkpoints include (a) the DNA damage checkpoints that recognize and respond to DNA damage, and (b) the DNA replication checkpoint that monitors the fidelity of copying DNA. http://www.eishinoguchi.com/
DNA maintenance checkpoint;
(a) DNA damage checkpoint
DNA damage checkpoints ensure the fidelity of genetic information both by arresting cell cycle progression and facilitating DNA repair pathways. Studies on many different species have uncovered a network of proteins that form the DNA damage checkpoints. Central to this network are protein kinases of ATM/ATR family known as Tel1/Mec1 in budding yeast and Tel1/Rad3 in fission yeast4. These kinases sense DNA damages and phosphorylate number of proteins that regulate cell cycle progression and DNA repair pathways
(b) DNA replication checkpoint
Accurate replication of the millions or billions of DNA base pairs in a eukaryotic genome is a remarkable achievement. This accomplishment is even more astonishing when one considers for DNA synthesis are rarely ideal. Damaged template, protein complexes bound to DNA, and poor supply of dNTPs are among the many obstacles that must be overcome to replicate genome. All of these situations can stall replication forks. Stalled forks pose grave threats to genome integrity because they can rearrange, break, or collapse through disassembly of the replication complex. The pathways that respond to replication stress are signal transduction pathways that are conserved across evolution. Atop the pathways are also ATM/ATR family kinases. These kinases together with a trimeric checkpoint clamp (termed 9-1-1 complex) and five-subunit checkpoint clamp loader (Rad17-RFC2-RFC3-RFC4-RFC5) senses stalled replication forks and transmit a checkpoint signal. One of major functions of replication checkpoint is to prevent the onset of mitosis by regulating mitotic control proteins such as Cdc25. But perhaps the most important activity of replication checkpoint is to stabilize and protect replication forks. The protein kinase Cds1 (human Chk2homolog; in human, Chk1 is a functional Cds1 homolog) is a critical effector of the replication checkpoint in the fission yeast Schizosaccharomycs pombe. Cds1 is required to prevent stabilization of replication fork in cells treated with hydroxyurea (HU), a ribonucleotide reductase inhibitor that stalls replication by depleting dNTPs. In the budding yeast Saccharomyces cerevisiae, a failure to activate Rad53 (Chk2 homolog) is associated with collapse and regression of replication forks and gross chromosomal rearrangements in cells treated with HU
Accurate duplication of eukaryotic genome is a challenging task, given that environment of cell growth and division is rarely ideal. Cells are constantly under the stress of intrinsic and extrinsic agents that cause DNA damage or interference with DNA replication. To cope with these assaults, cells are equipped with DNA maintenance checkpoints 3 to arrest cell cycle and facilitate DNA repair pathways. DNA maintenance checkpoints include (a) the DNA damage checkpoints that recognize and respond to DNA damage, and (b) the DNA replication checkpoint that monitors the fidelity of copying DNA.
Replication fork protection complex (FPC)
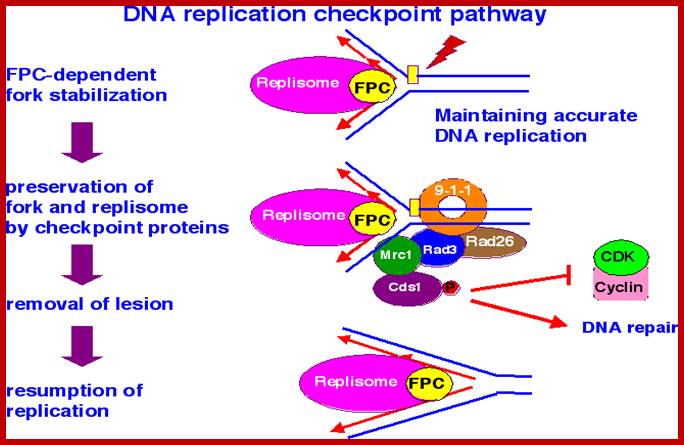
The DNA replication checkpoint stabilizes replication forks that have stalled at DNA adducts and other lesions that block DNA polymerases. In the absence of DNA replication checkpoint, stalled forks are thought to collapse, creating strand break that threatens genome stability and cell viability . Therefore, discovering how cells cope with aberrant replication forks is essential for understanding mechanisms of genome maintenance. The Chk1 and Chk2/Cds1 checkpoint kinases, which are key mediators of DNA damage and DNA replication checkpoints, are thought to be involved in cancer development. We found the Swi1 protein is required for survival of replication fork arrest and effective activation of Chk2 kinase in fission yeast. Swi1 forms tight complex with Swi3 protein and moves with replication forks.Swi1-Swi3 complex is also important for proficient DNA replication even in the absence of agents that cause genotoxic stress, creating single-strand DNA gaps at replication forks. These results led us to propose Swi1-Swi3 define a replication fork protection complex (FPC) that stabilizes replication forks in a configuration that is recognized by replication checkpoint sensors. Interestingly, Tof1protein (Budding yeast Swi1 homolog) has been reported to have similar functions. Tof1 is also involved in Rad53 (Chk2 homolog) activation and travels with replication fork 18, 19. Tof1 is needed to restrain fork progression when DNA synthesis is inhibited by HU indicating that Tof1 is required for coordination of DNA synthesis and replisome (replication machinery) movement. http://www.eishinoguchi.com/
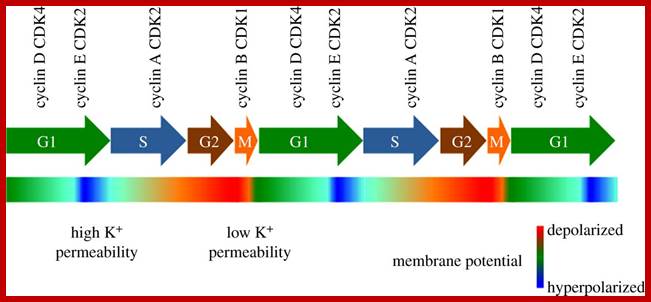
Potassium channels in cell cycle and cell proliferation; at specific stages of cell cycle specific cyclin-CDKs operate;
Here, authors summarize the possible mechanisms underlying the importance of potassium channels in cell-cycle control and briefly review some of the identified channels that illustrate the multiple ways in which this group of proteins can influence cell proliferation and modulate cell-cycle progression; http://rstb.royalsocietypublishing.org/
P53, cell cycle control and apoptosis: Implications for cancer; http://link.springer.com/Tumors frequently have decreased cell death as a primary mode of increased cell proliferation; loss of apoptosis is the signal for cancer; P53 protein is an example of a gene product which affects both cell cycle progression and apoptosis. The ability of p53 overexpression to induce apoptosis may be a major reason why tumor cells frequently disable p53 during the transformation process. Loss of apoptosis during tumor development may also result in tumor cell resistance to anti-neoplastic therapies which kill tumor cells by apoptosis.
The Rb protein;
Retinoblastoma is an eye cancer; this cancer is due to the non-functioning or absence of this gene called Rb protein. RB is a tumour suppressor, which plays a pivotal role in the negative control of the cell cycle and tumour progression. It has been shown that Rb protein (pRb) is responsible for a major G1 checkpoint, blocking S-phase entry and cell growth. The retinoblastoma family: RB1 (human) mapping to 13q14.2; includes three members, Rb/p105, p107 and Rb2/p130, collectively referred to as 'pocket proteins'. RB protein is 928a.a long-mol.wt-110kDa. The mutant causes Retinoblastoma in human eyes a dreaded tumour. The retinoblastoma a tumour suppressor gene product, known as Rb or pRb, regulates differentiation, apoptosis and cell cycle control by coordinating the cell cycle at G1/S with transcriptional machinery that includes the heterodimeric E2F family. The pRb protein represses gene transcription, required for transition from G1 to S phase, by directly binding to the transactivation domain of E2F and by binding to the promoter of these genes as a complex with E2F. pRb represses transcription also by remodelling chromatin structure through interaction with proteins such as hBRM, BRG1, HDAC1 and SUV39H1, which are involved in nucleosome remodelling, histone acetylation/deacetylation and methylation, respectively. Loss of pRb functions may induce cell cycle deregulation and so lead to a malignant phenotype. Gene inactivation of pRB through chromosomal mutations is one of the principal reasons for retinoblastoma tumour development. Functional inactivation of pRb by viral oncoprotein binding is also shown in many neoplasia such as cervical cancer, mesothelioma and AIDS-related Burkitt's lymphoma.
The mammalian cell cycle machinery: Upon receiving stimulatory influences from either cytokines or growth factors, mammalian cells undergo a regulated cell cycle progression. A typical cell cycle is divided into G1, S, G2 and M phases. During progression through the G1 phase, cells pass through 'a restriction point ( R )' where cells can leave the cell cycle and enter a reversible quiescence phase (G0), repair damaged DNA, if any and re-enter the cell cycle upon completion of repair, depending on the available mitogenic stimulus. In contrast, in the event of an un-repairable DNA damage scenario, cells can exit the cell cycle at the restriction point and undergo apoptosis. Also, at the restriction point, cells can undergo differentiation, which is an irreversible exit from the cell cycle. During conditions of low mitogenic stimulus, cells exit the cell cycle at the restriction point and undergo quiescence, which can be a reversible process wherein cells can re-enter the cell cycle upon availability of appropriate mitogenic stimulation. In case of cells with a limited life-span, the cell cycle exit is irreversible senescence followed by apoptosis. Cells committed to pass through the cell cycle progress through the S-phase (where DNA synthesis occurs), G2 and M-phase (where cells undergo mitosis). Every phase of the cell cycle is under regulatory influences of different cell cycle proteins. Thus, cyclin D-Cdk4/6 proteins are activated in early G1-phase where the cyclin D-Cdk4/6 assembly and optimal activity requires the p21 and p27 KIP/CIP family proteins. The p21 and p27 proteins inhibit the kinase activities of Cdk2 and cdc2 kinases, complexed with either cyclin E or cyclin A, during late G1, S, G2 and M-phases. In contrast, the p21 and p27 proteins are essential for proper assembly and subsequent activation of the cyclin D-Cdk4/6 complex. The INK4 (inhibitors of Kinase 4) proteins, p15, p16, p18 and p19, inhibit cyclin D-Cdk4/6 kinase activities. The concomitant result of cyclin D-Cdk4/6 activation is phosphorylation of the retinoblastoma (pRb) family or proteins. Phosphorylation of pRb by the cyclin D-Cdk4/6 complex results in a release of histone deacetylase (HDAC) from pRb proteins. Relief of the E2F proteins which occurs after subsequent phosphorylation of pRb by Cyclin E-Cdk2 leads to release of bound E2F resulting in increased expression of genes required for DNA synthesis during S-phase. The pRb proteins are maintained in their hyperphosphorylated state upon subsequent phosphorylation by cyclin A-Cdk2 and cyclin B-Cdk2 or Cdc2 (Cdk1) complexes. This maintenance of pRb proteins in hyper-phosphorylated state precludes the necessity for additional mitogenic stimulation to ensure completion of the cell cycle. Finally, the return of the pRb proteins back to their hypo-phosphorylated states, which leads to re-sequestration of the HDAC and E2F proteins, takes the cell cycle back to ground state. Further cell cycle turns will require resumption of mitogenic stimulation and re-activation of the cell cycle kinases. https://www.bioscience.org/.
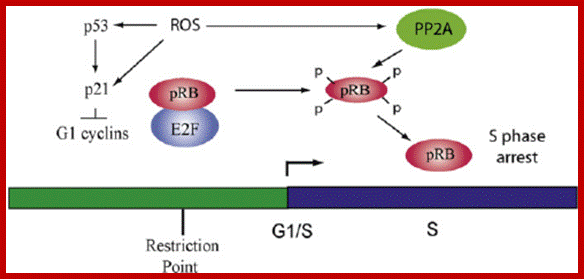
pRB is an effector of cell cycle arrest in response to ROS. In actively cycling cells mitogenic signaling through Ras in G2 controls the decision to continue cycling after mitosis. As cells progress through G1, ROS in fl uence p53-dependent and p53- independent mechanisms that control expression of p21, which in turn regulates the activity of G1 cyclin/CDKs required for phosphorylation of pRB and activation of the S- phase transcription factor E2F. Once cells are in S phase, exposure to ROS or RNS induces the dephosphorylation of pRB by the phosphatase PP2A, which results in S-phase arrest that is not dependent on the DNA-damage response. PP2A apparently differs from other phosphatases in that it is activated by calcium in response to oxidants, whereas oxidation inactivates phosphatases like PTPIB and PTEN. https://www.researchgate.net/
Molecular mechanisms underlying RB protein function:
RB protein; https://en.wikipedia.org
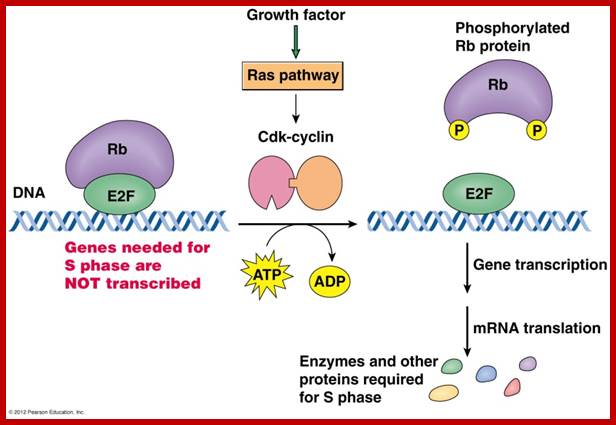
:
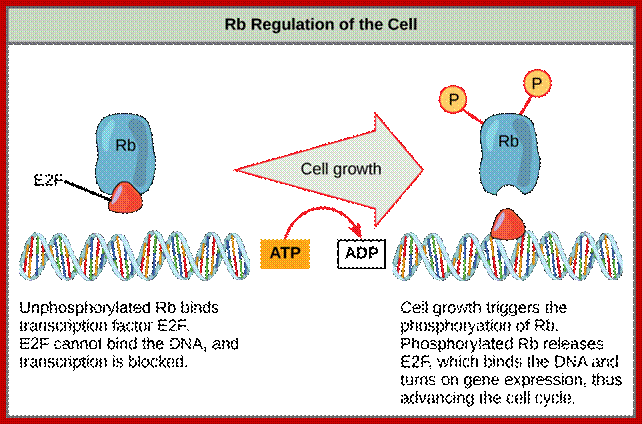
RB and cell cycle progression; C. Giacinti and A Giordano-http://www.nature.com/
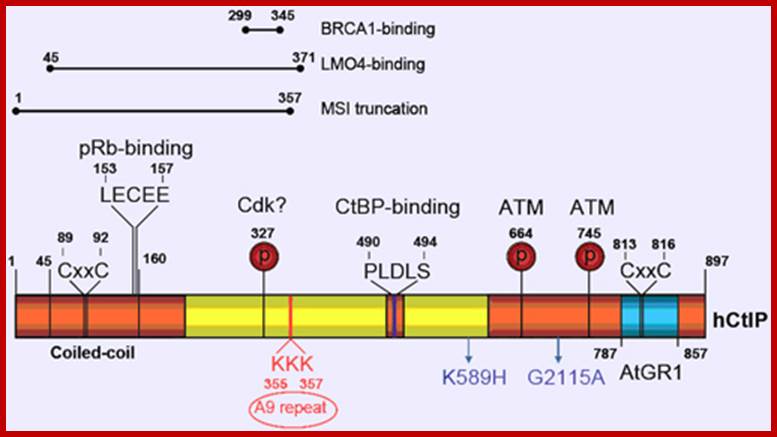
http://atlasgeneticsoncology.org/

http://www.nature.com/
Post-translational modifications have an important role in the regulation of RB function. With a few exceptions, RB phosphorylation (P) results in inactivation, transcriptional derepression and cell cycle progression. RB is phosphorylated by several different kinases, including cyclin-dependent kinases (CDKs) and checkpoint kinase 2 (CHK2). Phosphorylation controls RB interactions with other proteins. This modification typically occurs outside structured domains (see the figure) and promotes conformational transitions from disordered to ordered RB structures that mask protein-binding surfaces. Different kinases show preferences for particular phosphorylation sites, and discrete phosphorylation events induce specific structural changes. However, it remains uncertain whether, and in what context, differentially phosphorylated isoforms of RB exist in the cell.
Acetylation (Ac) and methylation (Me) sites have been identified in disordered sequences towards the RB carboxy-terminal domain (RBC). In contrast to phosphorylation, these modifications occur in response to signals, such as DNA damage and differentiation, which correlate with RB activation and repression of gene expression. Acetylation occurs on Lys873 and Lys874, which are located within the cyclin-docking sequence, and results in reduced phosphorylation, probably through kinase inhibition. Methylation on Lys873 and Lys810 by SET-domain methyltransferases similarly results in RB hypo phosphorylation. SET and MYND domain-containing 2 (SMYD2) methylates Lys860, which results in the recruitment of the transcriptional repressor lethal malignant brain tumour-like 1 (L3MBTL1). The reader is referred to other reviews for more details on these and other emerging post-translational modifications on RB and their roles in the regulation of function.
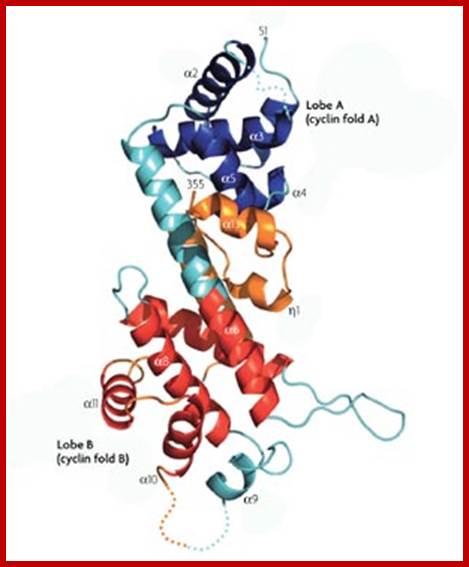
The Rb protein is a tumour suppressor, which plays a pivotal role in the negative control of the cell cycle and in tumour progression. It has been shown that Rb protein (pRb) is responsible for a major G1 checkpoint, blocking S-phase entry and cell growth. The retinoblastoma family includes three members, Rb/p105, p107 and Rb2/p130, collectively referred to as 'pocket proteins'. The pRb protein represses gene transcription, required for transition from G1 to S phase, by directly binding to the transactivation domain of E2F and by binding to the promoter of these genes as a complex with E2F. pRb represses transcription also by remodelling chromatin structure through interaction with proteins such as hBRM, BRG1, HDAC1 and SUV39H1, which are involved in nucleosome remodelling, histone acetylation/deacetylation and methylation, respectively. Loss of pRb functions may induce cell cycle deregulation and so lead to a malignant phenotype. Gene inactivation of pRB through chromosomal mutations is one of the principal reasons for retinoblastoma tumour development. Functional inactivation of pRb by viral oncoprotein binding is also shown in many neoplasia’s such as cervical cancer, mesothelioma and AIDS-related Burkitt's lymphoma. http://www.ncbi.nlm.nih.gov/
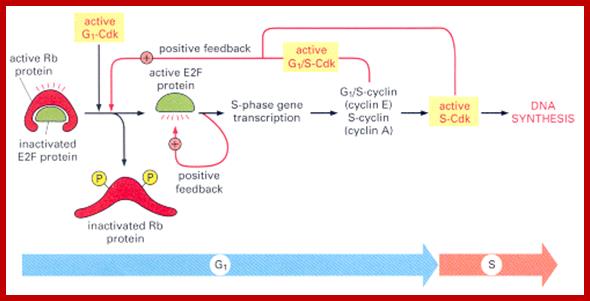
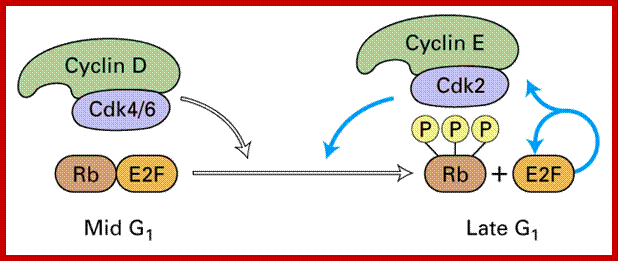
http://www.chegg.com/
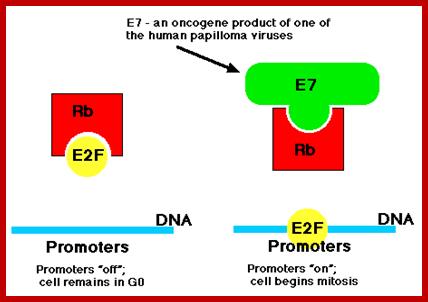
http://atlasgeneticsoncology.org/
Once inside the cells of their host, these viruses synthesize a protein designated E7 and another designated E6. Of the >30 strains of HPV that infect humans, several, especially HVP-16 and HPV-18, have been implicated as a risk factor for cervical cancer and also cancers of the throat. Their E7 protein binds to the Rb protein preventing it from binding to the host transcription factor E2F.
Free E2F binds to the promoters of genes (like c-myc) that cause the cell to enter the cell cycle (right). Thus, this version of E7 is an oncogene product. The E6 protein binds the p53 protein targeting it for destruction by proteasomes and thus removing the block on the host cell's entering the cell cycle. Although the figure shows the "off" promoters as empty, it is now clear that being "off" involves both
- the absence of activators of transcription and
- the presence of repressors of transcription.
G1- , depending on internal and external conditions, it can delay G1, enter a quiescent state known as G0, or proceed past the restriction point. The decision to commit to a new round of cell division occurs when the cell activates cyclin-CDK-dependent transcription which promotes entry into S phase.
During early G1, the transcriptional repressors Rb (retinoblastoma), p107 and p130, known as pocket proteins, bind to the E2F transcription factors to prevent G1-to-S transition. https://en.wikipedia.org,
Rb binds and represses activator E2F transcription factors (E2F1-3), while p107 and p130 bind E2F4 and E2F5 to form complexes which repress transcription of G1-to-S promoting factors. Upon the decision to progress past the G1 checkpoint, cyclin D levels rise, and cyclin D forms a complex with CDK4 and CDK6, which in turn phosphorylate the pocket proteins. Phosphorylation of the pocket proteins causes the release of their bound targets, thereby relieving the repression of the E2F1-3 activators and translocating repressors E2F4 and E2F5 from the nucleus to the cytoplasm. This results in the transcriptional activation of downstream targets, which promote the G1-to-S transition, including another cyclin, known as cyclin E, which forms a complex with CDK2. The formation of the cyclin E-CDK2 complex then promotes a positive feedback loop which creates an “all or nothing” switch from which the cell cannot return. Following entry to S-phase and initiation of DNA replication, S-phase cyclin A, a transcriptional target of E2F1-3, forms a complex with CDK2 which phosphorylates E2F1-3 and prevents its ability to bind to DNA, thus forming a negative feedback loop. In another negative feedback loop, E2F1-3 promote the transcription of E2F6-8, which in turn repress G1-S transition.
When DNA is damaged; it to delay or halt the cell cycle in G1, The rapid response involves phosphorylation events that initiate with either kinase ATM (Ataxia telangiectasia mutated) or ATR (Ataxia Telangiectasia and Rad3 related), which act as sensors, depending on the type of damage. These kinases phosphorylate and activate the effector kinases Chk2 and Chk1, respectively, which in turn phosphorylate the phosphatase Cdc25A, thus marking it for ubiquitination and degradation. As Cdc25A activates cyclin-E-CDK2 complex by removing inhibitory phosphates from CDK2, in the absence of Cdc25A, cyclin E-CDK2 remains inactive, and the cell remains in G1.
To maintain the arrest, another response is initiated, by which Chk2 or Chk1 phosphorylate p53, a tumor suppressor, and this stabilizes p53 by preventing it from binding Mdm2, a ubiquitin ligase which inhibits p53 by targeting it for degradation. The stable p53 then acts a transcriptional activator of several target genes, including p21, an inhibitor of the G1-to-S promoting complex cyclin E-CDK21. In addition, another mechanism by which p21 is activated is through the accumulation of p16 in response to DNA damage. p16 disrupts cyclin D-CDK4 complexes, thus causing the release of p21 from the complexes, which leads to the dephosphorylation and activation of Rb, which allows Rb to bind and inhibit E2F1-3, thus keeping the cell from transitioning to S phase. Recently, some aspects of this model has been disputed.
p21 is a potent cyclin-dependent kinase inhibitor (CKI). The p21 (CIP1/WAF1) protein binds to and inhibits the activity of cyclin-CDK2, -CDK1, and -CDK4/6 complexes, and thus functions as a regulator of cell cycle progression at G1 and S phase. In addition to growth arrest, p21 can mediate cellular senescence. One of the ways it was discovered was as a senescent cell-derived inhibitor.wikipedia.org.
The expression of this gene is tightly controlled by the tumour suppressor protein p53, through which this protein mediates the p53-dependent cell cycle G1 phase arrest in response to a variety of stress stimuli.
Commitment to enter S-phase occurs through sequential phosphorylation of Rb by Cyclin D-CDK4/6 and Cyclin E-CDK2 that dissociates the HDAC-repressor complex, permitting transcription of genes required for DNA replication. In the presence of growth factors, Akt can phosphorylate FoxO1/3, which inhibits their function by nuclear export, thereby allowing cell survival and proliferation. Importantly, a multitude of different stimuli exert checkpoint control, including TGF-β, DNA damage, replicative senescence, and growth factor withdrawal. Collectively, ubiquitin/proteasome-dependent degradation and nuclear export are mechanisms commonly used to effectively reduce the concentration of cell cycle control proteins. Importantly, Cyclin D1/CKD4/6 complexes are explored as therapeutic targets for cancer treatment as researchers have found this checkpoint to be invariantly deregulated in human tumours. https://www.cellsignal.com/
The Rb protein is a tumour suppressor, which plays a pivotal role in the negative control of the cell cycle and in tumour progression. It has been shown that Rb protein (pRb) is responsible for a major G1 checkpoint, blocking S-phase entry and cell growth. The retinoblastoma family includes three members, Rb/p105, p107 and Rb2/p130, collectively referred to as 'pocket proteins'. The pRb protein represses gene transcription, required for transition from G1 to S phase, by directly binding to the transactivation domain of E2F and by binding to the promoter of these genes as a complex with E2F. pRb represses transcription also by remodeling chromatin structure through interaction with proteins such as hBRM, BRG1, HDAC1 and SUV39H1, which are involved in nucleosome remodeling, histone acetylation/deacetylation and methylation, respectively. Loss of pRb functions may induce cell cycle deregulation and so lead to a malignant phenotype. Gene inactivation of pRB through chromosomal mutations is one of the principal reasons for retinoblastoma tumour development. Functional inactivation of pRb by viral oncoprotein binding is also shown in many neoplasia such as cervical cancer, mesothelioma and AIDS-related Burkitt's lymphoma. http://www.nature.com/
The Rb gene is functionally inactivated in most human neoplasms either by direct mutation/deletion, such as in retinoblastoma, osteosarcoma and small-cell lung carcinoma, or indirectly through altered expression/activity of upstream regulators; responsible to check G1 phase b lock entr into S-phase. The Rb gene family includes three members, Rb/p105, p107 and Rb2/p130, collectively referred to as 'pocket proteins' over expression of them arrest cell at G1
Moreover, the interaction between the pRb family proteins and the E2F family transcription factors plays a central role in governing cell cycle progression and DNA replication by controlling the expression of cell cycle E2F-dependent genes. In addition, pRb recruits chromatin remodeling factors such as histone deacetylase 1 (HDAC1) SWI/SNF factors, Polycomb group proteins or methyltransferase that act on the nearby surrounding nucleosome structure.
The most convincing evidence of the importance of pRb in cellular differentiation comes from studies of Rb knockout mice, where the disruptions of the Rb gene cause death by day 14 of gestation, associated with defects in the development of the hematopoietic system and central nervous system
P53: p53 is also called as TP53- tumor protein;
P53 is the guardian of the Genome. Tumor suppressor protein- Identified by Arnold Levine, David Lane and Williams Old from Princeton University. P53 gene is located in chromosomes 17p13.1, the protein is 393aa long, it has four domains- Transcription activator domain, DNA sequence specific binding domain, tetramer forming domains, domain that recognizes DNA damage and misaligned Base pair or single stranded DNA.
More than 50% of abnormal growth to cancer are due to lack or mutated P53. Functions-growth arrest by activating genes responsible for DNA repair perse; if all fail, finally induce apoptosis as a last resort. Major regulator of P53 is Mdm2. Functions in growth arrest by activating genes responsible DNA repair. Major regulator of P53 is Mdm2. P53 protein’s N-terminal domain perform transcriptional activation, Growth arrest-by p21, Gadd45, and 14-3-3s, DNA repair; p53R2. It is also associated with Bax, Apaf-1, PUMA and NOx A. P53 unphosphorylated gets degraded by ubiquitin mediated proteasome complex; phosphorylated 53 form Mdm2 p53 complex. P53 regulates the expression of these inhibitory proteins to induce growth arrest. P53 with also acts with Bax and Apaf1 and activates Caspase9 leading to apoptosis. But p53 is continuously produced and degraded in the cell. If one of the 2 p53 genes is mutated, persons get Li-Fraumeni syndrome. With only one copy of p53 in a diploid cell likely to get tumors in the later stage. In vitro injection of p53 into p53 deficient cells cause rapid cell deaths of cancer cells.
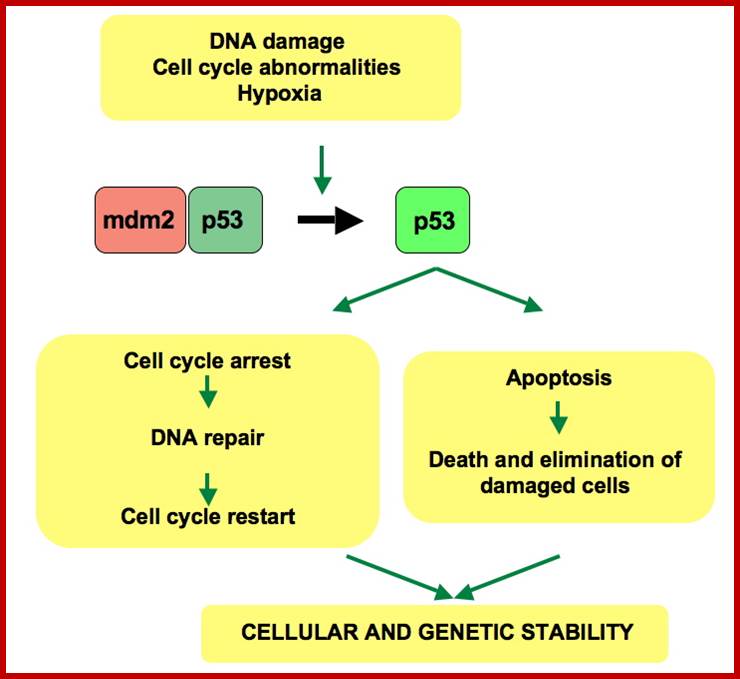
p53 pathway: In a normal cell, p53 is inactivated by its negative regulator, mdm2. Upon DNA damage or other stresses, various pathways will lead to the dissociation of the p53 and mdm2 complex. Once activated, p53 will induce a cell cycle arrest to allow either repair or survival of the cell or apoptosis to discard the damaged cell. How p53 makes this choice is currently unknown. https://en.wikipedia.org/wiki/P53
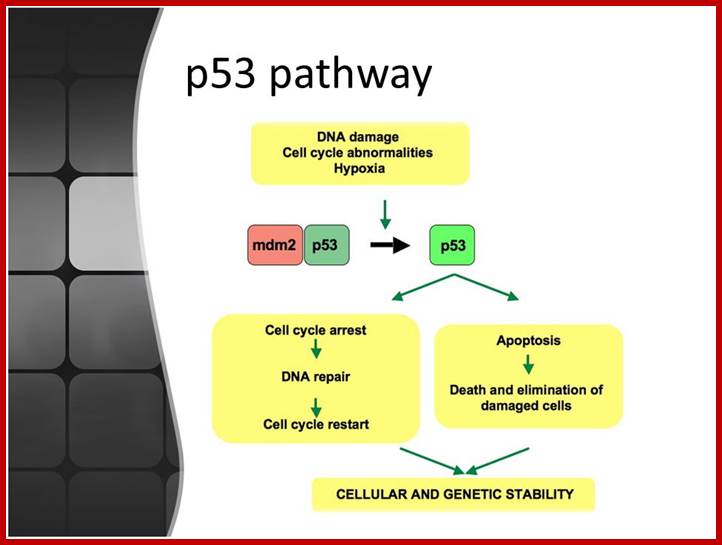
Heterochromatin silencing at p53 gene targets by a small viral protein; http://slideplayer.com/
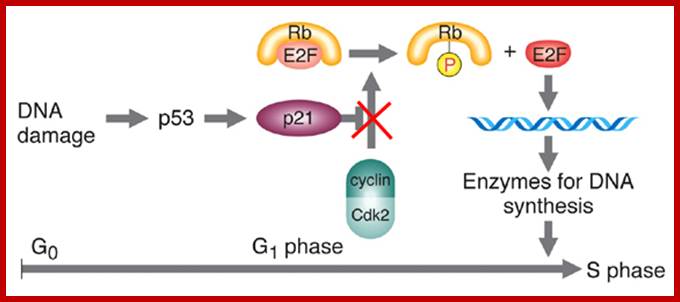
http://www.bio.miami.edu/dana/
Other tumor suppressor mutations involve problems with positive regulation of apoptosis (e.g., p53).
- Mutations of p53 are associated with many different types of cancers.
- p53 is thus considered a TUMOR SUPPRESSOR GENE
- Normal P53 protein is a transcription factor activated in response to DNA damage.
- prevents progression of cell cycle in the presence of damaged DNA
- can induce apoptosis under some circumstances
- Mutant p53 cannot induce apoptosis, and cannot halt the cell cycle in the presence of damaged DNA.
- This results in an increase in the overall frequency of mutations
- If some of these mutations occur in proteins regulating proliferation and apoptosis, CANCER will result.
- Carcinogenesis of p53 causes cancer indirectly, as it simply causes an elevated rate of mutation, increasing the chances of carcinogenic utations.
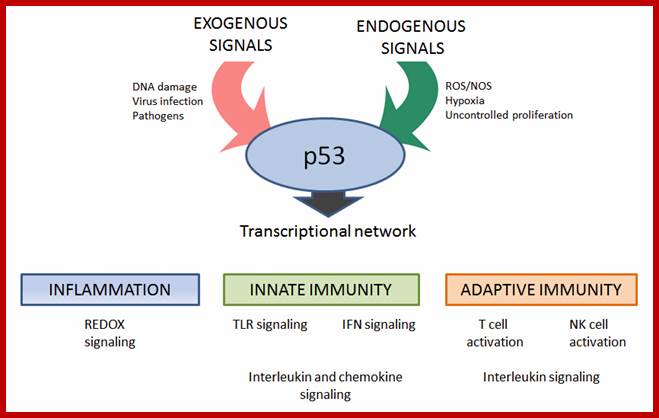
Modulation of immune responses by the tumor suppressor p53; http://www.biodiscoveryjournal.co.uk/
The immune system is a collection of biological processes whose tasks in preventing disease include identification and destruction of pathogens and tumor cells. Given the broad diversity in p53 controls and functions, it is not surprising that p53 touches multiple aspects of immunity. For example, DNA damage can trigger p53 responses that help orchestrate clearance of damaged cells via the innate immune system, which can influence tumor suppression. In addition p53 is up-regulated at sites of inflammation, likely due to the appearance of reactive oxygen species (ROS) that might damage DNA and proteins. The seminal work of Xue and his colleagues demonstrated the functional relationship between p53 and the immune system in a mouse liver carcinoma model containing a “switchable” p53. In this study they showed that p53 and the immune system can cooperate to promote tumor clearance. They found that p53-dependent tumor regression was related to induction of a tumor cell-senescence program, associated differentiation, up-regulation of pro-inflammatory cytokines and activation of innate immune response.
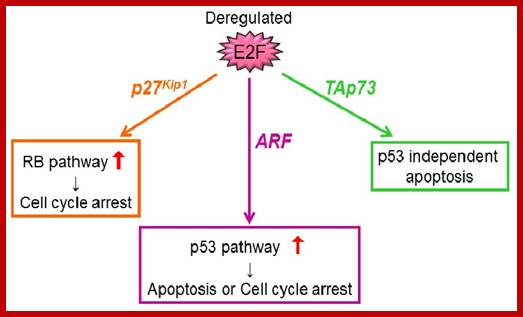
To Grow, or not to Grow, Stop or Die? – Novel Tumor-Suppressive Mechanism Regulated by the Transcription Factor E2F;http://www.intechopen.com/
P53- mol wt 43.7Kda a phosphoprotein, it shows 53Kda in gel, is due the presence of many prolines which slows the migration in SDS gel. Human 53 encode 15 isoforms (p53isoforms). Nearly 50% of the cancers is due to nonfunctional p53.
The gene is located on chromosome 17(17p13.1), size is ~10kb; The coding sequence contains five regions showing a high degree of conservation in vertebrates, predominantly in exons 2, 5, 6, 7 and 8, but the sequences found in invertebrates show only distant resemblance to mammalian TP53. A common polymorphism involves the substitution of an arginine for a proline at codon position 72. Many studies have investigated a genetic link between this variation.
1. an acidic N-terminus transcription-activation domain (TAD), also known as activation domain 1 (AD1), which activates transcription factors. The N-terminus contains two complementary transcriptional activation domains, with a major one at residues 1–42 and a minor one at residues 55–75, specifically involved in the regulation of several pro-apoptotic genes.
2. activation domain 2 (AD2) important for apoptotic activity: residues 43-63.
3. proline rich domain important for the apoptotic activity of p53 by nuclear exportation via MAPK: residues 64-92.
4. central DNA-binding core domain (DBD). Contains one zinc atom and several arginine amino acids: residues 102-292. This region is responsible for binding the p53 co-repressor LMO3.
5. nuclear localization signaling domain, residues 316-325.
6. homo-oligomerization domain (OD): residues 307-355. Tetramerization is essential for the activity of p53 in vivo.
7. C-terminal involved in downregulation of DNA binding of the central domain: residues 356-393.
A tandem of nine-amino-acid transactivation domains (9aaTAD) was identified in the AD1 and AD2 regions of transcription factor p53. KO mutations and position for p53 interaction with TFIID shown below. The competence of the p53 transactivation domains 9aaTAD to activate transcription as small peptides was reported. Mutation DBD makes it more potent in cancer, for it is non active.
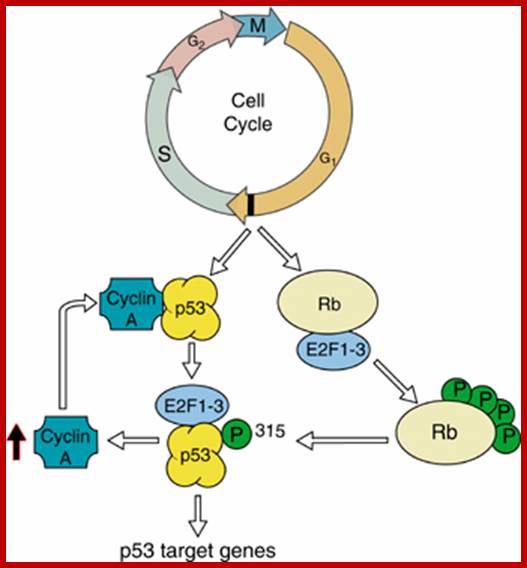
http://dpuadweb.depauw.edu/; Cell cycle control of p53 as cell cycle check point protein functions by E2F family members; p53 get phosphorylated at ser315 by cell cycle associate kinases. This provokes E2F1-3 to bind to p53 displacing CyclinA interaction and inducing p53 transcriptionally active/competent; A W Braithwaite, G Del Sal and X Lu; http;//www.nature.com
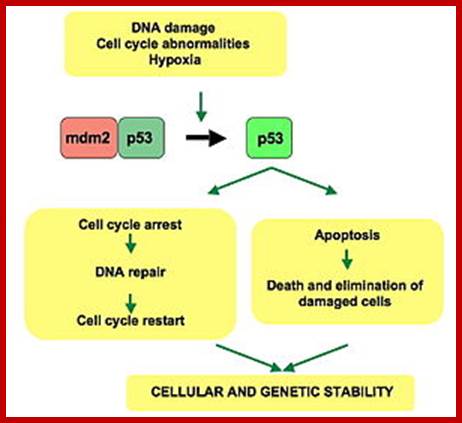
p53 pathway: In a normal cell, p53 is inactivated by its negative regulator, mdm2. Upon DNA damage or other stresses, various pathways will lead to the dissociation of the p53 and mdm2 complex. Once activated, p53 will induce a cell cycle arrest to allow either repair and manke survive of the cell or apoptosis to discard the damaged cell. How p53 makes this choice is currently unknown. https://en.wikipedia.org/
p53 has many mechanisms of anticancer function and plays a role in apoptosis, genomic stability, and inhibition of angiogenesis. In its anti-cancer role, p53 works through several mechanisms:
· It can activate DNA repair proteins when DNA has sustained damage. Thus, it may be an important factor in aging.
· It can arrest growth by holding the cell cycle at the G1/S regulation point on DNA damage recognition (if it holds the cell here for long enough, the DNA repair proteins will have time to fix the damage and the cell will be allowed to continue the cell cycle).
· It can initiate apoptosis (i.e., programmed cell death) if DNA damage proves to be irreparable.
Activated p53 binds DNA and activates expression of several genes including microRNA miR-34a, WAF1/CIP1 encoding for p21 and hundreds of other down-stream genes. p21 (WAF1) binds to the G1-S/CDK (CDK4/CDK6, CDK2, and CDK1) complexes (molecules important for the G1/S transition in the cell cycle) inhibiting their activity.
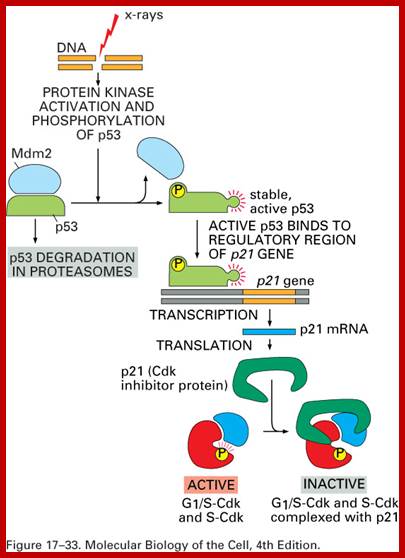
http://www.pha.jhu.edu/~ghzheng
When p21 (WAF1) is complexed with CDK2, the cell cannot continue to the next stage of cell division. A mutant p53 will no longer bind DNA in an effective way, and, as a consequence, the p21 protein will not be available to act as the "stop signal" for cell division. Studies of human embryonic stem cells (hESCs) commonly describe the non-functional p53-p21 axis of the G1/S checkpoint pathway with subsequent relevance for cell cycle regulation and the DNA damage response (DDR). Importantly, p21 mRNA is clearly present and upregulated after the DDR in hESCs, but p21 protein is not detectable. In this cell type, p53 activates numerous microRNAs (like miR-302a, miR-302b, miR-302c, and miR-302d) that directly inhibit the p21 expression in hESCs.
p53 becomes activated in response to myriad stressors, including but not limited to DNA damage (induced by either UV,IR, or chemical agents such as hydrogen peroxide), oxidative stress, osmotic shock, ribonucleotide depletion, and deregulated oncogene expression. Recent research has also linked the p53 and RB1 pathways, via p14ARF, raising the possibility that the pathways may regulate each other.
A ubiquitin specific protease, USP7 (or HAUSP), can cleave ubiquitin off p53, thereby protecting it from proteasome-dependent degradation. This is one means by which p53 is stabilized in response to oncogenic insults. USP42 has also been shown to deubiquitinate p53 and may be required for the ability of p53 to respond to stress.
Phosphorylation also allows for binding of transcriptional coactivators, like p300 and PCAF, which then acetylate the carboxy-terminal end of p53, exposing the DNA binding domain of p53, allowing it to activate or repress specific genes. Deacetylase enzymes, such as Sirt1 and Sirt7, can deacetylate p53, leading to an inhibition of apoptosis. Some oncogenes can also stimulate the transcription of proteins that bind to MDM2 and inhibit its activity. P53 interacts with at least 100 or more proteins like AIMP2, NUMB, NFkB, p16 and others.
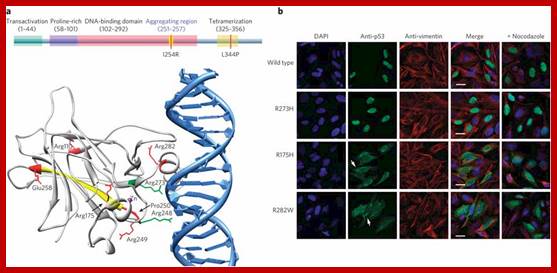
The schematic domain structure of p53 is shown in the upper panel. An aggregation-prone sequence sits in the DNA-binding domain, spanning the residues 251 to 257. Mutations that can inhibit aggregation (I254R) or abolish tetramerization (L344P) are labeled in red. The structure of p53 DNA-binding domain is shown in the lower panel. The structural mutations (R110P, R175H, R248Q, R249S and R282W) and the contact mutations (R248W and R273H) are labeled in red and green, respectively. The aggregation-prone sequence is shown in yellow. (PDB ID: 1TUP, image generated by VMD software, http://www.ks.uiuc.edu/). (b) The Do-1 antibody revealed intense punctate cytoplasmic staining (white arrows) for the structural mutants R175H and R282W but not the contact mutant R273H and the wild-type p53. The cytoplasmic aggregates were caged by collapsed vimentin, and the treatment with nocodazole produces a diffuse cytoplasmic staining for the aggregating mutants. Scale bars: 10 μm. https://en.wikipedia.org/wiki/P53
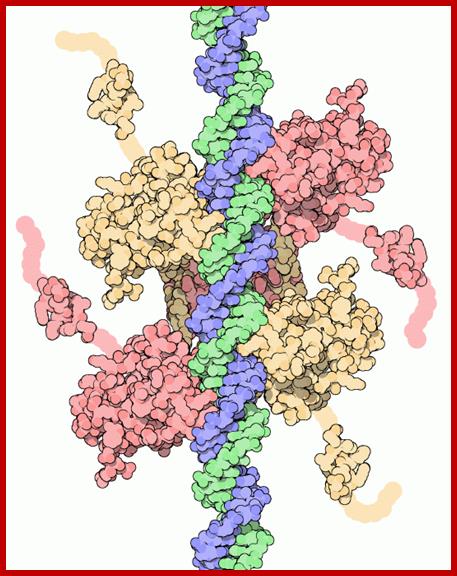
P53- https://pdb101.rcsb.org/motm/31
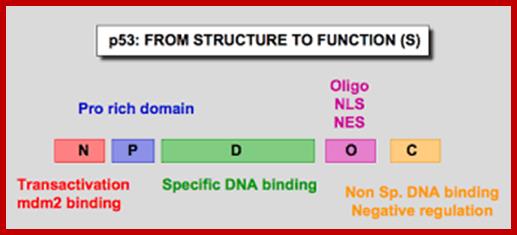
P53.Free.fr- Domains
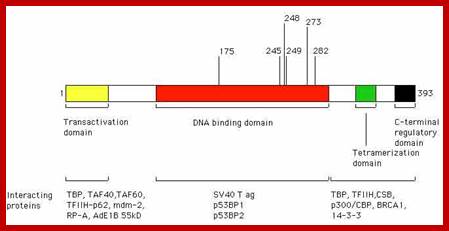
Frontiers in Biosciences-April 2000; http://www.bioscience.org/
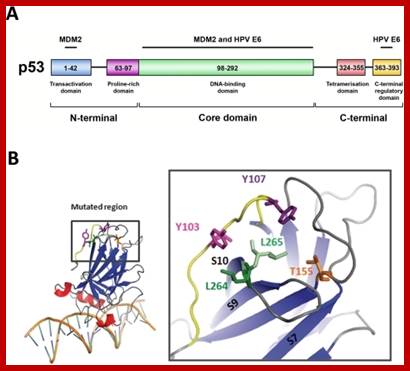
P53 domains;https://openi.nlm.nih.gov

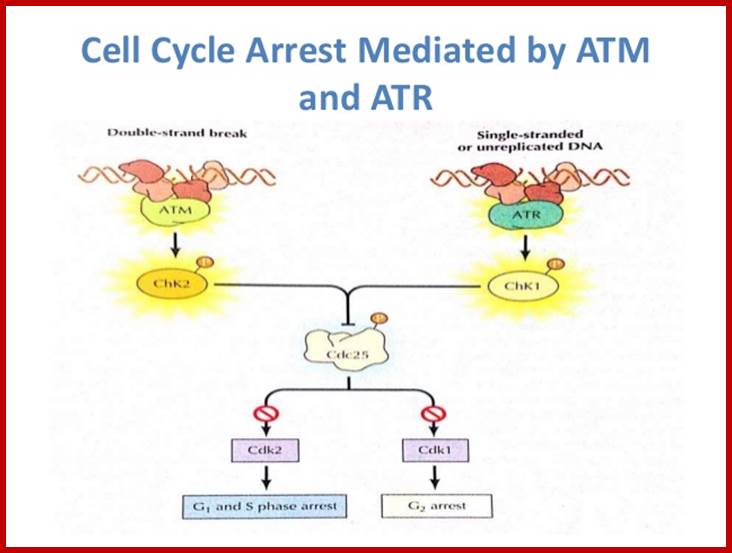
Shubankar;http://image.slidesharecdn.com/
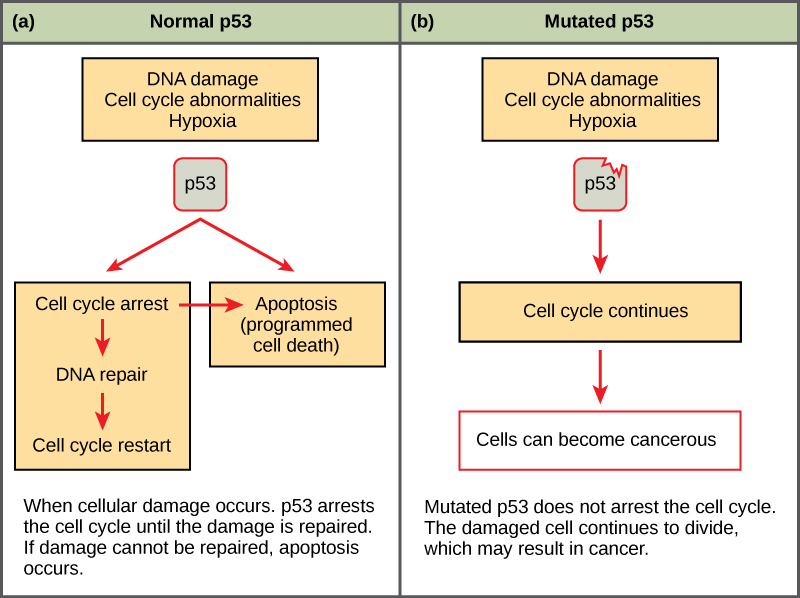
(a) The role of p53 is to monitor DNA. If damage is detected, p53 triggers repair mechanisms. If repairs are unsuccessful, p53 signals apoptosis. (b) A cell with an abnormal p53 protein cannot repair damaged DNA and cannot signal apoptosis. Cells with abnormal p53 can become cancerous. (credit: modification of work by Thierry Sousse) https://opentextbc.ca
CHEK proteins are CHEK point serine/threonine kinases;
CHEK2 protein ; https://en.wikipedia.org/wiki; https://en.wikipedia.org/wiki/CHEK2- Chek2
CHEK 2 is a tumor suppressor; CHK2 operates in an intricate network of proteins to elicit DNA repair, cell cycle arrest or apoptosis in response to DNA damage. Mutations to the CHEK2 gene have been linked to a wide range of cancers including breast cancer.
Structure of the CHEK1 protein: Based on PyMOL rendering of PDB; Checkpoint kinase 1, commonly referred to as Chk1 is an Serine/threonine-specific protein kinase that in humans, is encoded by the CHEK1 gene. Chk1 coordinates the DNA damage response (DDR) and cell cycle checkpoint response. Activation of Chk1 results in the initiation of cell cycle checkpoints, cell cycle arrest, DNA repair and cell death to prevent damaged cells from progressing through the cell cycle. Chk1 contains four Ser/Gln residues.[6] Chk 1 activation occurs primarily through the phosphorylation of the conserved sites, Ser-317, Ser-345 and less often at Ser-366. CHK1- performs DNA repair, cell cycle arrest; CHK1 is activated by ATR, CHK1 regulates M phase; G2-M transition, S- phase.
The protein encoded by this gene belongs to the Ser/Thr protein kinase family. It is required for checkpoint mediated cell cycle arrest in response to DNA damage or the presence of unreplicated DNA. This protein acts to integrate signals from ATM and ATR, two cell cycle proteins involved in DNA damage responses, that also associate with chromatin in meiotic prophase I. Phosphorylation of CDC25A protein phosphatase by this protein is required for cells to delay cell cycle progression in response to double-strand DNA breaks. Several alternatively spliced transcript variants have been found for this gene. [provided by RefSeq, Oct 2011]; CHEK1 (Checkpoint Kinase 1) is a Protein Coding gene. Diseases associated with CHEK1 include ataxia-telangiectasia and breast cancer. Among its related pathways are Gene Expression and Signaling by GPCR. GO annotations related to this gene include transferase activity, transferring phosphorus-containing groups and protein tyrosine kinase activity. An important paralog of this gene is SBK2.
CHK2 operates in an intricate network of proteins to elicit DNA repair, cell cycle arrest or apoptosis in response to DNA damage. Mutations to the CHEK2 gene have been linked to a wide range of cancers including breast cancer.
CHK2 operates in an intricate network of proteins to elicit DNA repair, cell cycle arrest orapoptosis in response to DNA damage. Mutations to the CHEK2 gene have been linked to a wide range of cancers including breast cancer. The serine/threonine kinase CHK2 is a key component of the DNA damage response. In human cells, following genotoxic stress, CHK2 is activated and phosphorylates >20 proteins to induce the appropriate cellular response, which, depending on the extent of damage, the cell type, and other factors, could be cell cycle checkpoint activation, induction of apoptosis or senescence, DNA repair, or tolerance of the damage. Recently, CHK2 has also been found to have cellular functions independent of the presence of nuclear DNA lesions. In particular, CHK2 participates in several molecular processes involved in DNA structure modification and cell cycle progression. In this review, we discuss the activity of CHK2 in response to DNA damage and in the maintenance of the biological functions in unstressed cells. These activities are also considered in relation to a possible role of CHK2 in tumorigenesis and, as a consequence, as a target of cancer therapy.
CHK2 kinase is active in the DNA damage response and beyond;
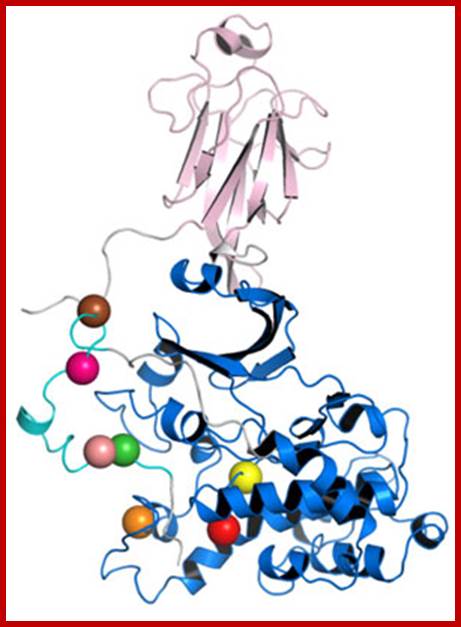
A
representative crystal structure of the Chk2 protein after computational
modelling and simulation of molecular dynamics; The coloured spheres represent
the locations of mutations, which BII scientists have found to be useful as a
prognostic marker for HG-SOC; www.a-star.edu.sg; http://jmcb.oxfordjournals.org/
{kind=link}
{kind=link}
Check point proteins are serine /threonine kinase2; CHEK2; www.Wikipedia.org
Chek2 is a tumor suppressor; CHK2 operates in an intricate network of proteins to elicit DNA repair, cell cycle arrest or apoptosis in response to DNA damage. Mutations to the CHEK2 gene have been linked to a wide range of cancers including breast cancer.
Chek1 Protein:
Chk1; https://en.wikipedia.org
Commonly referred to as Chk1 is a Serine/threonine-specific protein kinase that in humans is encoded by the CHEK1 gene. Chk1 coordinates the DNA damage response (DDR) and cell cycle checkpoint response. Activation of Chk1 results in the initiation of cell cycle checkpoints, cell cycle arrest, DNA repair and cell death to prevent damaged cells from progressing through the cell cycle. This protein acts to integrate signals from ATM and ATR, two cell cycle proteins involved in DNA damage responses, that also associate with chromatin in meiotic prophase I. Phosphorylation of CDC25A protein phosphatase by this protein is required for cells to delay cell cycle progression in response to double-strand DNA breaks. Several alternatively spliced transcript variants have been found for this gene.
Chk1 is a central component of genome surveillance pathways and is a key regulator of the cell cycle and cell survival. Chk1 is required for the initiation of DNA damage checkpoints and has recently been shown to play a role in the normal (unperturbed) cell cycle. Chk1 impacts various stages of the cell cycle including the S phase, G2/M transition and M phase. Accumulation of mutations and chromosomal aberrations is one of the hallmarks of cancer cells. This enhanced genetic instability is fueled by defects in the genome maintenance mechanisms including DNA repair and cell cycle checkpoint pathways. The emerging roles of the mammalian Chk1 and Chk2 kinases as key signal transducers within the complex network of genome integrity checkpoints, as candidate tumor suppressors disrupted in sporadic as well as some hereditary malignancies and as potential targets of new anticancer therapies.
Cell Cycle and its implication in Cancer Genetics:
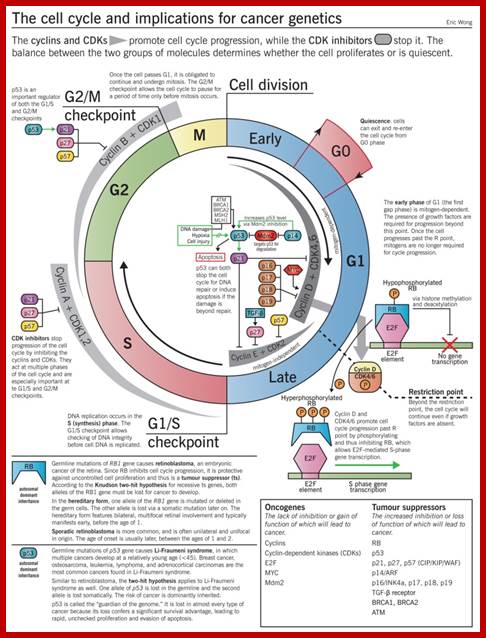
Cancer genetics: The regulator genes of the cell cycle are commonly mutated in cancers. Once cancer cells develop they can also induce more changes in genes so the cancer cells can grow and multiply uninhibited. Genes that stop cell cycle progression (an example of tumour suppressor genes) are often downregulated or missing, and genes that promote cell cycle progression (an example of proto-oncogenes) are often upregulated or made constitutively active. The end result is increased cell cycle progression, allowing tumour cells to proliferate without restraint. See the Cancer genetics chapter for details; http://www.pathophys.org; /https://in.pinterest.com, http://image.slidesharecdn.com/
· The accumulation of DNA damage in carcinogenesis usually triggers the intrinsic pathway to induce cell apoptosis.
· Thus, for neoplasia to form, the cell must evade the protective mechanism of apoptosis.
· BCL2, an anti-apoptotic protein, is commonly upregulated in cancers to protect against apoptosis.
- p53, a pro-apoptotic protein, is commonly downregulated in cancers to evade apoptosis, even in the face of irreparable DNA damage. http://www.pathophys.org/
The major changes in cellular biology that characterize cancer cells include: Self-sufficiency in growth signals: Insensitivity to anti-growth signals: Evasion of apoptosis: Limitless replicative potential: Sustained angiogenesis: Tissue invasion and metastasis:
P53, cell cycle control and apoptosis: Implications for cancer; http://link.springer.com/Tumors frequently have decreased cell death as a primary mode of increased cell proliferation; loss of apoptosis is the signal for cancer; P53 protein is an example of a gene product which affects both cell cycle progression and apoptosis. The ability of p53 overexpression to induce apoptosis may be a major reason why tumor cells frequently disable p53 during the transformation process. Loss of apoptosis during tumor development may also result in tumor cell resistance to anti-neoplastic therapies which kill tumor cells by apoptosis.
Check point protein induced Senescence, Necrosis and Apoptosis:
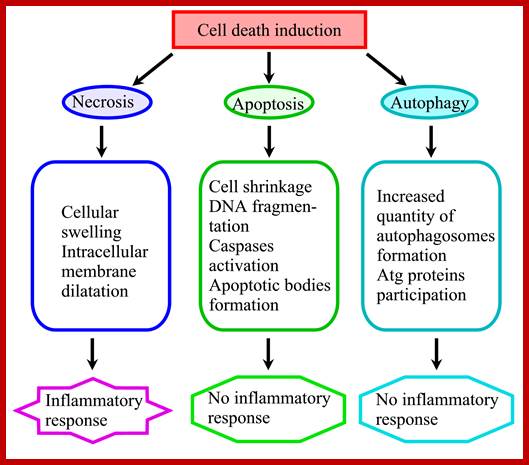
Steroid saponins and cell death; Diffrent types of programmed cell death; http://www.intechopen.com/
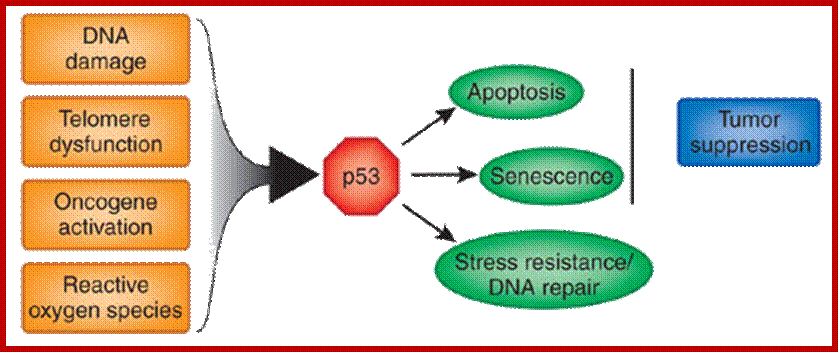
This page all started from a discussion I was having with my friend Jeff. about the role ATP depletion plays in triggering apoptosis and/or eliminating chemo resistance. I believe ATP depletion plays a supporting, not necessarily primary role in cancer treatment. I've been suggesting that constant usage of ATP depleting supplements inhibits apoptosis. This is an attempt to organize some of the reasons, methods, and tools I use to achieve cancer cell destinations.
New research on plant hormones like Jasmonates show that they deplete ATP and direct towards necrosis instead of apoptosis. I haven't been able to obtain Jasmonates, however something I do have is Paw. Paw which contains the most potent (ATP depleting) Annonaceous acetogenins. I also use Burdock and Dong Quaito push cancer cells over the nutrient starvation edge. http://www.ncbi.nlm.nih.gov/pubmed/18598079
p53--> p21-à STRESS RESISTANCE-.dna repairà stem cell dysfunctionà pro-AGING; STRESS RESISTANCE –TRANSIENTàSTEM CELL REPir and antiaging
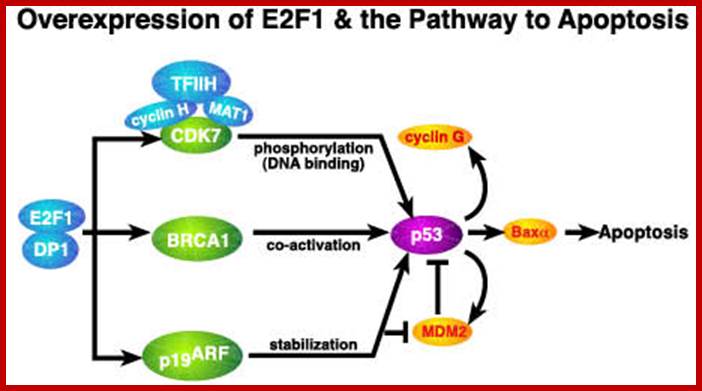
Over expression of E2F1 http://sciencepark.mdanderson.org/
Apoptosis and Necrosis:
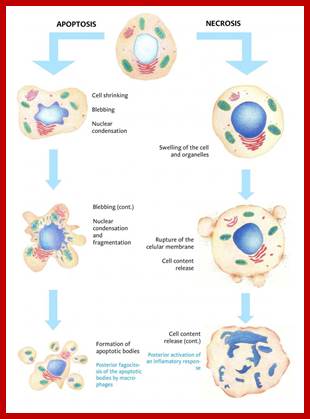
DNA damage— induces p53-that leads to Apoptosis; DNA damage also induces p53 that leads to aging and senescence; Morphological changes observed during apoptosis and necrosis; http://www.scienceinschool.org/
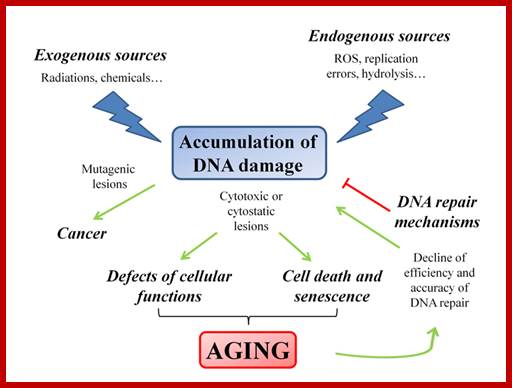
DNA damaged repairing leads to aging and senescence; Sara Nicolai et al http://archive.impactaging.com/

The hormones abscisic acid, ethylene, jasmonic acid and salicylic acid are accepted by most scientists as promoters of senescence, but at least one source lists gibberellins, brassinosteroids andstrigolactones as also being involved; https://en.wikipedia.org/

Final stage of leaf death with great coloration of leaves; http://www.biologyreference.com/