Proteins and Enzymes
Proteins:
Proteins are the products of structural genes, which on transcription get translated on mRNA template producing a polypeptide chain. This depending upon the amino acid sequences undergoes modifications at specific sites (amino acid residues), rarely splicing. The polypeptide chain organizes into secondary and tertiary structures. Most of the polypeptide chains based on their amino acid R groups first organize into helices (coiled structures) or extended chains called beta sheets. It is the sequences that ultimately determine the shape of proteins. Such secondary structured protein further fold into 3-D structures called tertiary structures by intra chain or inter-chain noncovalent bonds and covalent bonds. One or more monomers join with each other specifically to form polymer structures called quaternary structures. Total number of proteins found in human 10^14 cells and 667 tissue specific cells, is estimated to be 20,000 or more; this is based protein coding genes present in human genome; for 10^14 cells 20,000 proteins appears far less. (http://www.innovateus.net/) or but some say it can be more. Alternative splicing of mRNA from the same genes can generate more but different forms of proteins. But human genome contains protein coding genes about and codes for 20,000 (25,000 ?) proteins and 5000 gene for functional RNAs (http://www.nature.com/2014.); Mathias Wilhelm et al and Kim et al. Bernhard Kuster, a professor in Proteomics Techische University Monchen, in 2014 reported the existence of 18,097 proteins, what about the proteins generated by alternative splicing mRNAs. In humans ~95% of multi-exonic genes generated mRNAs. More than 60% to 70% of the gene products are generated due to alternative splicing. One gene multiple mRNAs is an accepted process. Different mRNAs can also be generated due to early site of transcription initiation and different sites of poly-A addition; but what is number of proteins added to the original proteomic analysis. E. coli produces approximately 5000 proteins. The number of Different proteins that can be generated by just using 20 different amino acid randomly attached to each other in a protein containing 100 amino acids; this combination can generate 20x100 or 20X^100 times; if the number of amino acids is 50 per a protein it can be 20 x 50 times 20x ^50 times.
http://sandwalk.blogspot.in/2015
Proteins can divide them into category- Fibrous (two), Globular (eleven) and Complex type with multiple components 9 more than four and each category contain different class of proteins. Among protein enzymes are different class of proteins but category of them is six. Basically, the kind of proteins are based on their functions, hormonal, Enzymatic, Structural, Defensive, Storage, Transport, Receptor and contractile. The fascinating aspects of proteins is that there is a specific relationship between amino acid sequences, nucleotide sequences in mRNA and nucleotide sequences in the genetic DNA; there is a flow of information from Gene-DNA to mRNA to Proteins. Understanding of Ramachandran’s plot is very important in understanding changes in protein structures.
Chemical Composition:
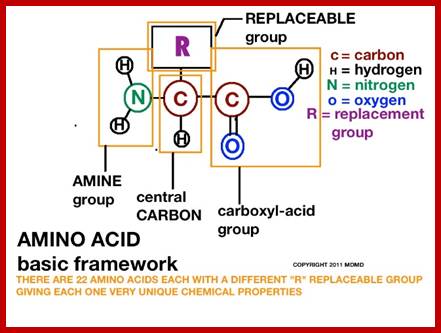
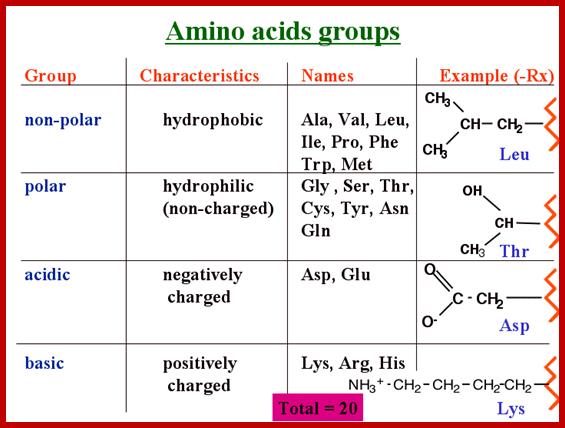
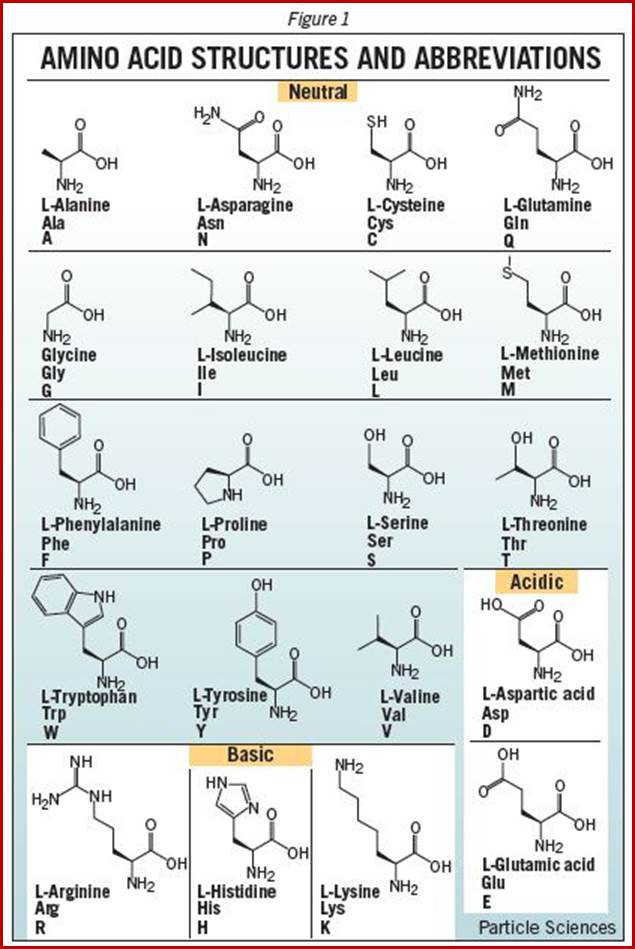
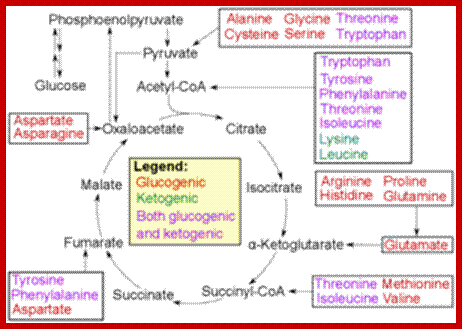
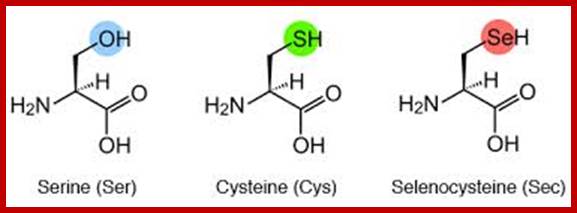
When proteins are hydrolyzed in the presence of strong acids, they yield amino acid residues. Living cells contain just twenty different kinds of amino acids, to which one more amino acid has to be added; that is Seleno-Cysteine; so, the total number of amino acids in biosystem is 21; which act as building blocks of protein chains; not only that amino acids are involved in basic cellular metabolism. However, different proteins contain different composition and different sequences of amino acid residues. A particular sequence generates a motif and several such motifs can generate a domain. There are several hundreds of different motifs and domains; and it is the combination of motifs and domains ultimately determines the structure and function of a protein. There is a distinct relationship between a motif and an exon, a coding sequence in the gene. Specific proteins post-translationally get modified with glycosylation, methylation and acetylation mostly and few others. The site and the numbers and the kind of modification differ from one protein to the other, it is genetically determined. It is the sequence of amino acids that ultimately determines the 3-D structure and the specific functions of the proteins.

www.particlesciences .com
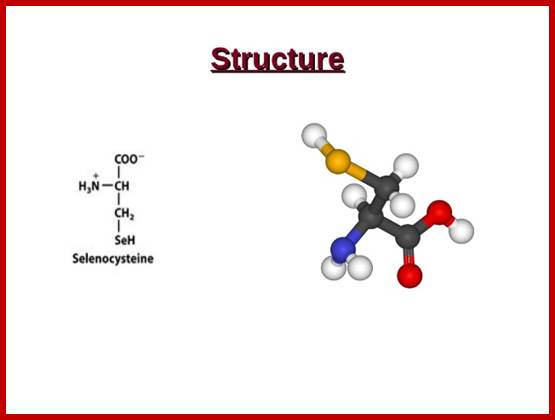
Selenocysteine;
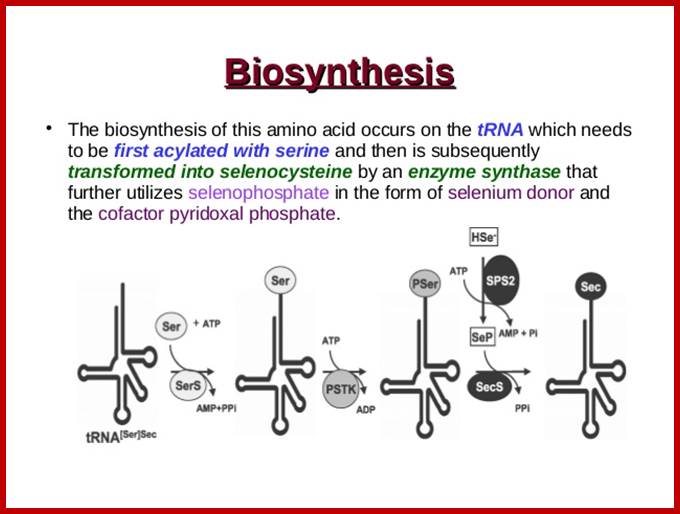
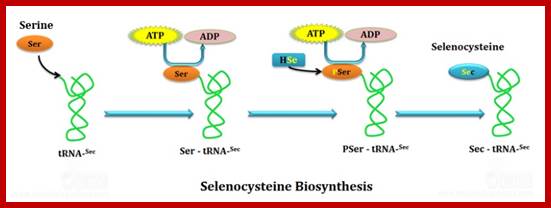
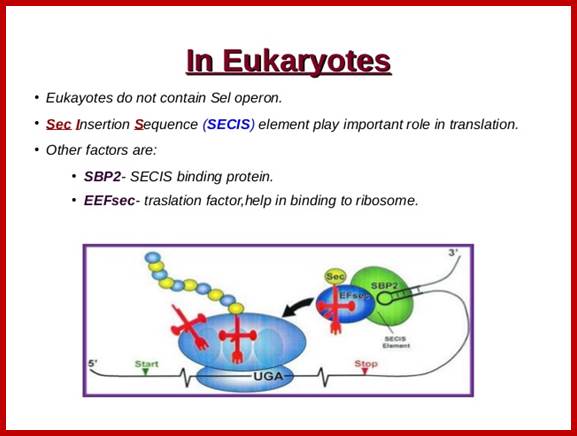
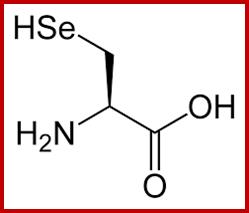
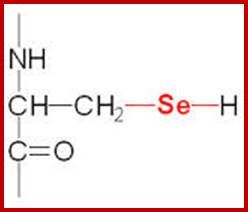
Selenocysteine-SelA (Sec or Se-cys) is the 21st amino acid. Discovered by Theresa Stadman (NIH); it is a building block of Seleno-proteins. SelA is synthesized when serine is added to tRNA. Then the serine is added with acetylation and then converted into cysteine and then to Selenocysteine; mechanism of conversion varies from archaea to bacteria to animal systems. It is an important component of several enzymes (for example glutathione peroxidases, tetraiodothyronine 5'deiodinases, thioredoxin reductases, formate dehydrogenases, glycine reductases, selenophosphate synthetase 1, methionine-R-sulfoxide reductase B1 (SEPX1), and some hydrogenases. Strangely selenocysteine has no genetic codon. Instead, it is encoded in a special way by a UGA codon, which is normally a stop codon. Such a mechanism is called translational recoding and its efficiency depends on the selenoprotein being synthesized and on translation initiation factors.
http://image.slidesharecdn.com/
http://image.slidesharecdn.com/
http://www.easybiologyclass.com/
http://image.slidesharecdn.com/
https://upload.wikimedia.org/wikipedia/commons/thumb/1/16/Amino_acid_catabolism_revised.png/300px-Amino_acid_catabolism_revised.png
Selenocysteine; www.themedicalbiochemistrypage.org
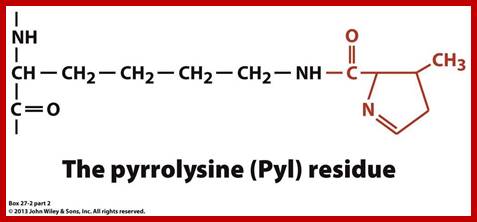
Pyrrolysine Pyrolysine-Pyl 1; It is considered as 22nd amino acid, it is named as amino acid amino acid; it is a proteinogenic amino acid. It is coded by UAG (actually UAG is terminator codon. Pyrrolysine is synthesized by joining of two lysines.
https://www.studyblue.com; http://image.slidesharecdn.com/
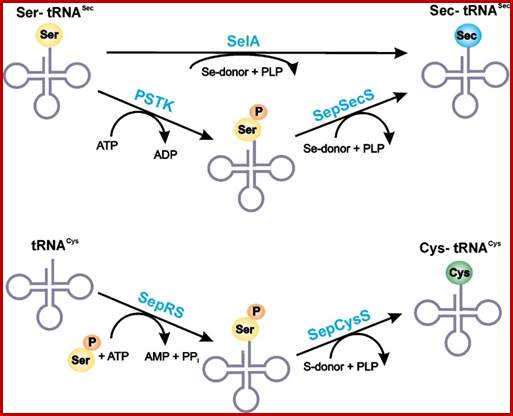
RNA-dependent conversion of phosphoserine forms selenocysteine in eukaryotes and archaea;
SelA route is bacterial and PSTK/SepSecS route is in Archaea and eukaryotes;www.pnas.org
SelA synthetic pathway; it differs from Archaea to bacteria and eukaryotic animals. www.spring8.or.jp
Linkage of a.a in the formation of proteins; Properties and their role:

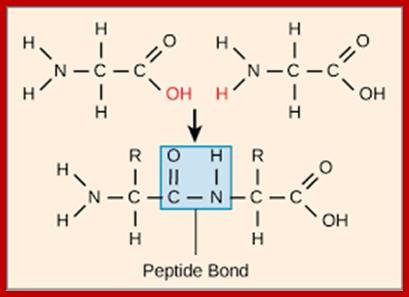
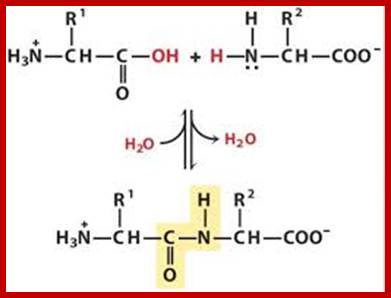
Peptide bond formation; www.Boundless,com
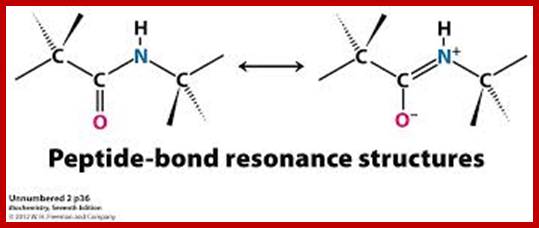
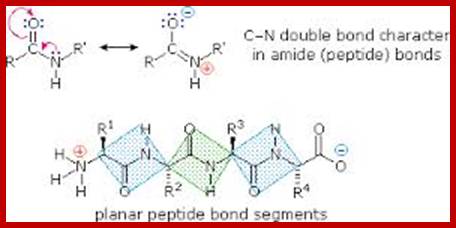
Peptide bon (C-N) bond formation; Fig-top, middle and bottom respectively; www.Studyblue.com; www. oregonstae.edu; www2chemistry.msu.edu
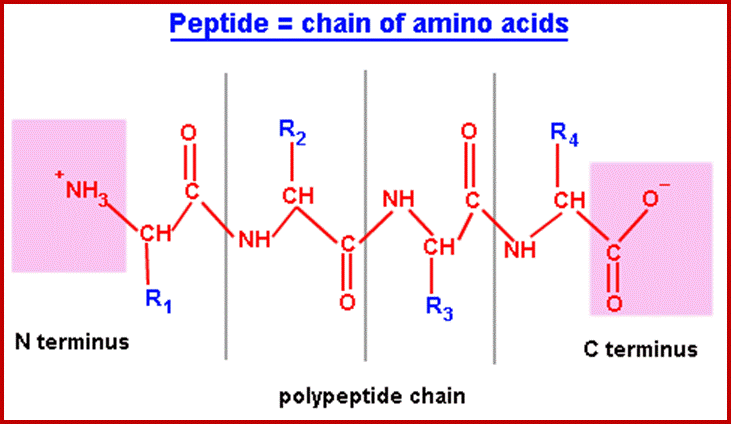
Peptide bonds are the amide bonds that link amino acids together to form peptides (short chains of amino acids) and proteins (longer polypeptide chains that fold into specific three-dimensional conformations with specific biological function).The C-N linkage between the carboxyl groups of one amino acid residue with an amino group of succeeding amino acid is called peptide bond. Based on the number of amino acid residues linked, proteins are called dipeptides, tripeptides and polypeptides.
A resonance structure, shown above, is invoked to explain the rigidity of the peptide group. Optimal electron delocalization (π-bonding) within the peptide group depends upon the planar arrangement of these atoms. The partial double-bond character of the peptide bond is reflected in the intermediate length of the peptide bond relative to single and double carbon-nitrogen bonds
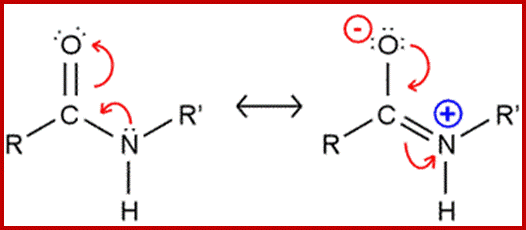
The peptide bond typically has a length of 1.32-1.33 Å (1 Å = 10−10 m), while the C–N bond in amines is typically ~1.49 Å, and the C=N bond of an imine is ~1.27 Å. guweb2.gonzaga.edu
“O=C – NH” is the C-N bond or Peptide Bond; on either side of the Peptide bond, one finds C-alpha of Amino acid residues which are linked. swissmodel.expasy.org
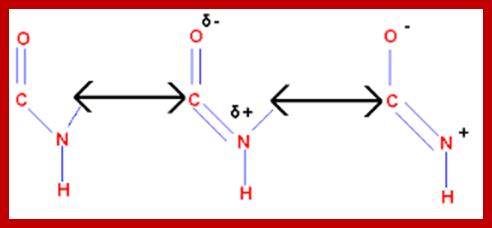
The peptide C-N bond can have alternate single or double bond, exchange with C=O; www.chemistry.tutorvista.com
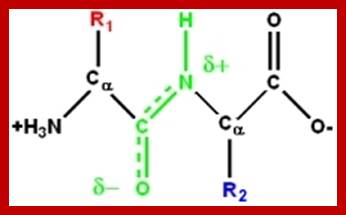
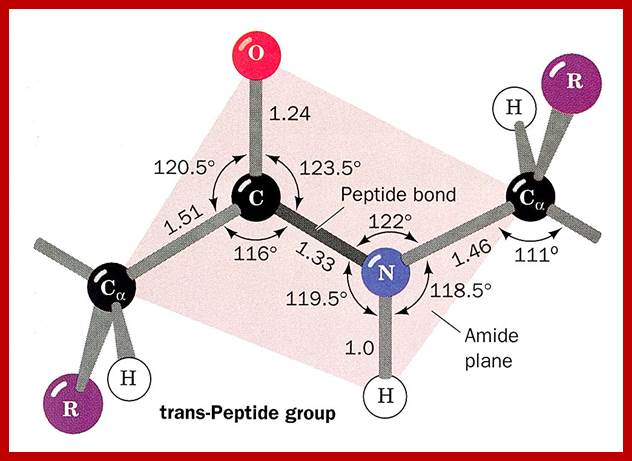
The planar peptide bond; Note that the oxygen and hydrogen atoms are on the opposite sides of the C-N bond. This is trans-configuration.); Linus Pauling and Robert Corey demonstrated that the α-Carbons of adjacent amino acids are separated by three covalent bonds, arranged Cα -C - N - Cα. The planar C-N bond is rigid; C-N bond in a peptide is planar, somewhat shorter than amide bonds (1.32 Å or 0.132nm); the double bond between C=O shifts toward C=N and back and forth dynamically. www.mikeblaber.org
www.imgarcade.com
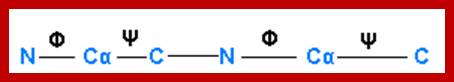
- The protein backbone can be described in terms of the phi, psi and omega torsion angles of the bonds: The phi angle is the angle around the -N-CA- bond (where 'CA' is the alpha-carbon)
- The psi angle is the angle around the -CA-C- bond
- The omega angle is the angle around the -C-N- bond (i.e. the peptide bond); http://www.bioinf.org.uk/
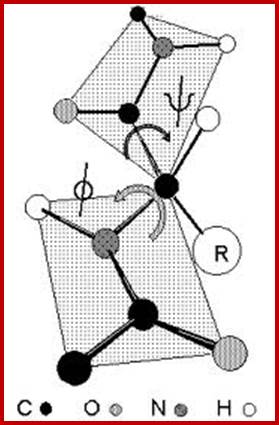
The rigidity of the peptide bond limits the number of arrangements that Pauling's models could fit without distorting bonds or forcing atoms closer than van der Waals radii would allow. Without this constraint, the peptide would be free to adopt so many structures that no single consistent pattern would emerge. By reducing the degrees of freedom, a well-defined set of states emerges. www.chembio.uoguelph.ca
Peptide bond linking two amino acid residues, planar and rigid, but the alpha carbon is can go through rotation. biologicalphysics.iop.org
www.imgarcade.com/1/polypeptide-polymer
Structural Features:
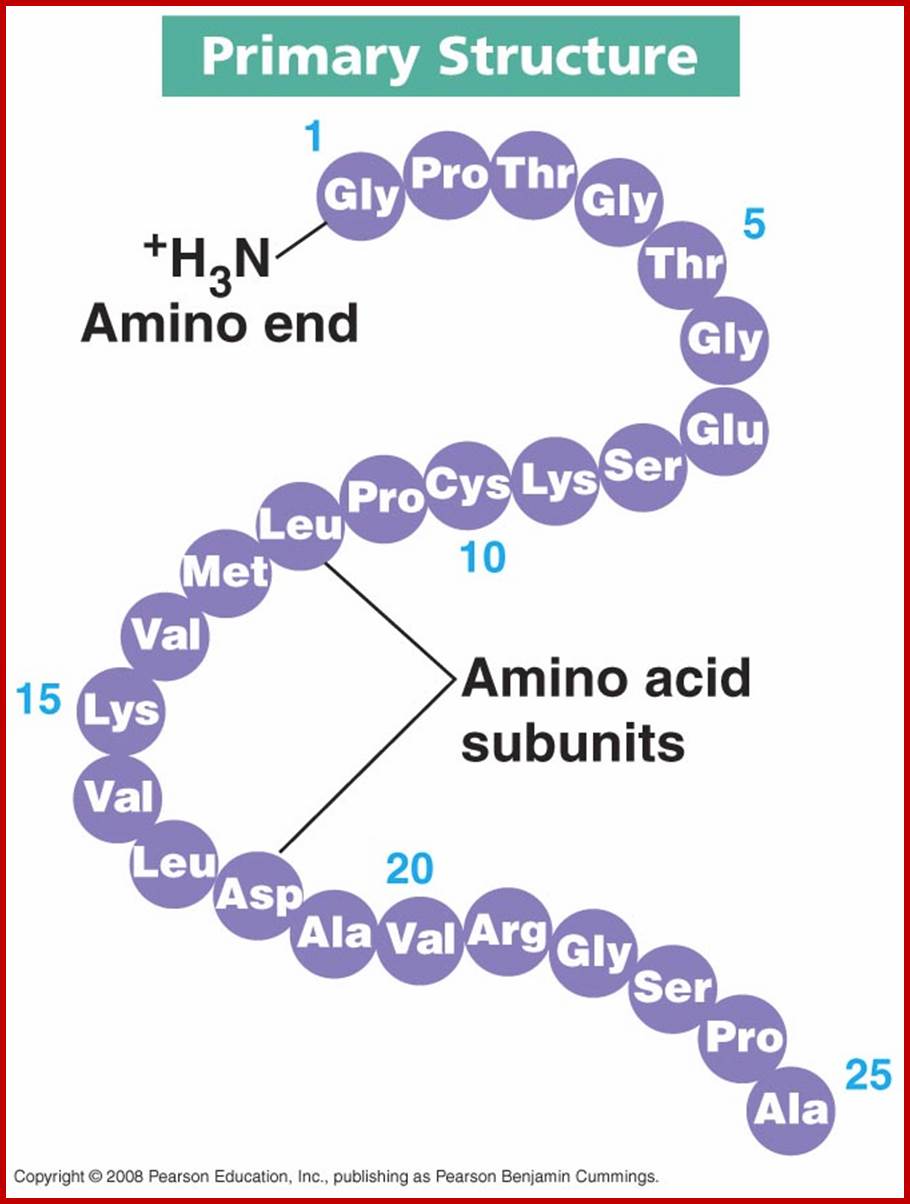
Primary structure:
Polypeptides chains assume different structural forms- such as primary and secondary, tertiary and quaternary forms; this is based on their amino acid sequence and protein-protein interaction domains.
The information that can be obtained from the primary structure is the composition and sequential arrangement of amino acid residues, in the polypeptide chain. This can be established by subjecting the putative protein into ‘finger printing’ techniques, which involves the identification of amino acid sequence of the polypeptide chain. The primary structure ultimately determines the other structural forms of proteins.
https://kimberlybiochemist.wordpress.com
Proteins are the polymers of amino acid residues held by peptide linkages. Next to them, carbohydrates, proteins and fatty acids are the major organic constituents of the cell. They are in every conceivable structure and functional components of the cell. They perform a wide variety of functions. Their role in cellular metabolism is so pervasive, if one of the proteins is missing, it will be fatal for the survival of the cell. In humans it will be expressed as a disease.
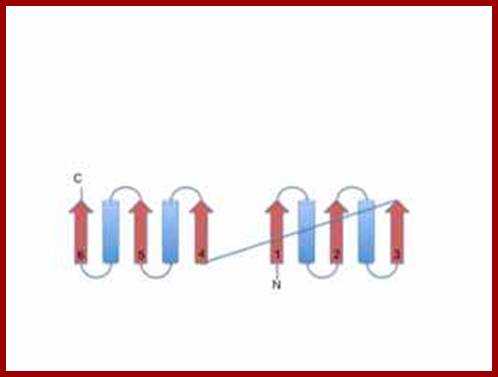
The definitions of primary, secondary, tertiary, and quaternary structure are as above and the definitions of the other levels of structure are-Super secondary structure is the next level up from secondary structure and involves the association of secondary structures; also known as structural motifs.
Motif: Please note that the term 'motif' is often used in other contexts. For example, a motif can be either a structural or a sequence motif:
- A structural motif is the arrangement of atoms in 3-D space to produce a particular structural pattern eg. Alpha helix, beta sheet.
- A sequence motif is a particular pattern in the sequence of amino acids or nucleotides. A structural or sequence motif may or may not have a particular function associated with it.
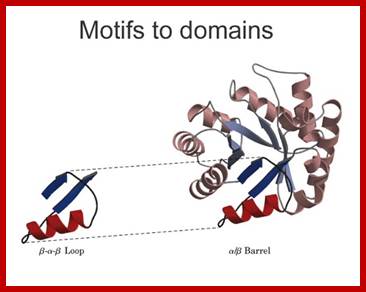
Motifs to domain; http://xray.bmc.uu.se/
Generally an average polypeptide chain of mol.wt of 12 KD-36 KD consists of 110-330 amino acid residues.
Sometimes certain compounds other than amino acids are added to proteins at amino acid in endoplasmic reticulum (mostly); here amino acid group are modified or added with ex. carbohydrates, lipids, sulfates, nucleotides, metals, etc. Accordingly, the proteins are called glycoproteins, lipoproteins, sulfoproteins, nucleoproteins and metalloproteins. The above said components are added only after the synthesis of polypeptide chains. Some polypeptide chains are made up of Inteins and Exteins; and Inteins are removed by self-splicing activity by the border amino acid residues; which are similar to mRNA splicing. Furthermore, a protein may be made up of a single polypeptide chain, called monomer or it may contain more than one polypeptide chains called dimers, trimers, polymers, etc. Such polymers have protein-protein interacting surfaces; they come in different forms and shapes.
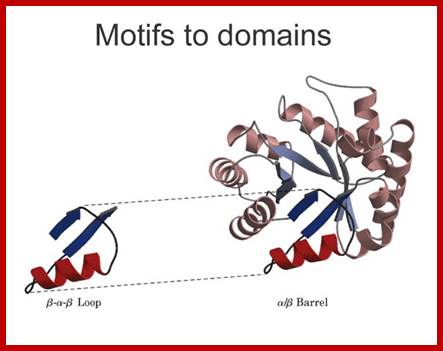
http://xray.bmc.uu.se/
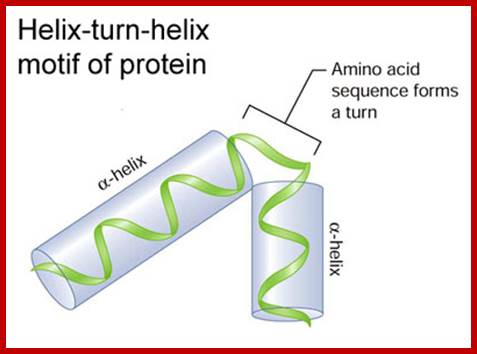
http://carrot.mcb.uconn.edu/
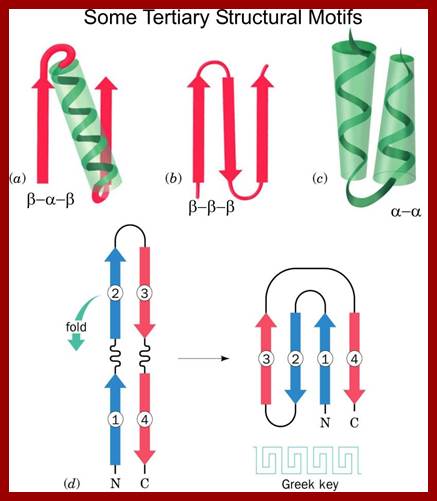
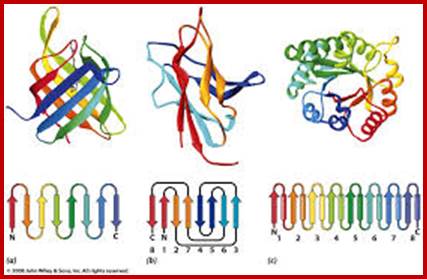
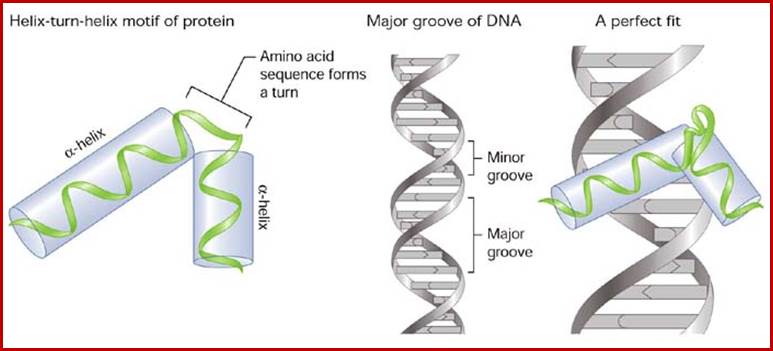
Helix turn helix motif, helix loop helix motifs, EF-hand calcium binding motifs; Few structural motifs; http://chemistry.ewu.edu/
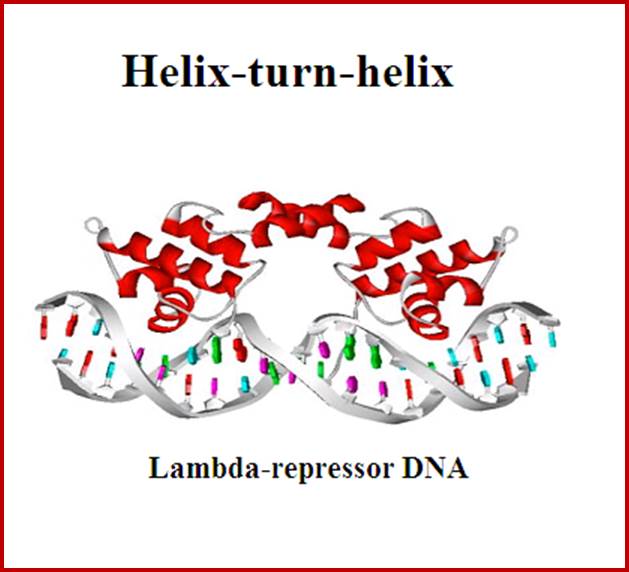
Hekix-turn Helix motif- a DNA binding domain; www.swift.cmbi.ru.nl
Levels of protein structures; 3-D structural organization of beta and alpha helix segments; https://www.ebi.ac.uk
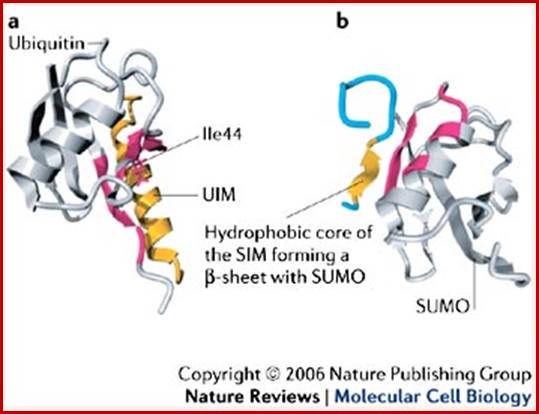
Comparison of the related folds of ubiquitin and SUMO. Motifs- a sequence leading to secondary structure like helix can act As motif as shown above.
Ubiquitin-interacting Motif (UIM) (yellow color) of vacuolar sorting protein-27 bound to ubiquitin; The PIASX protein inhibitor of activated STAT SUMO interacting Motif (yellow color) bound to SUMO; http://www.nature.com/
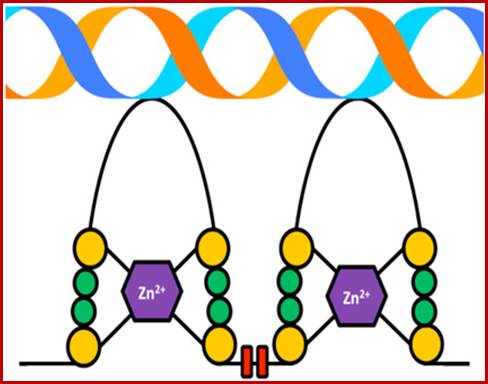
Zinc Finger Motif fits nicely into DNA helical groove; the two tandem repeat Zn motifs constitute the LIM domain; http://www.sigmaaldrich.com/
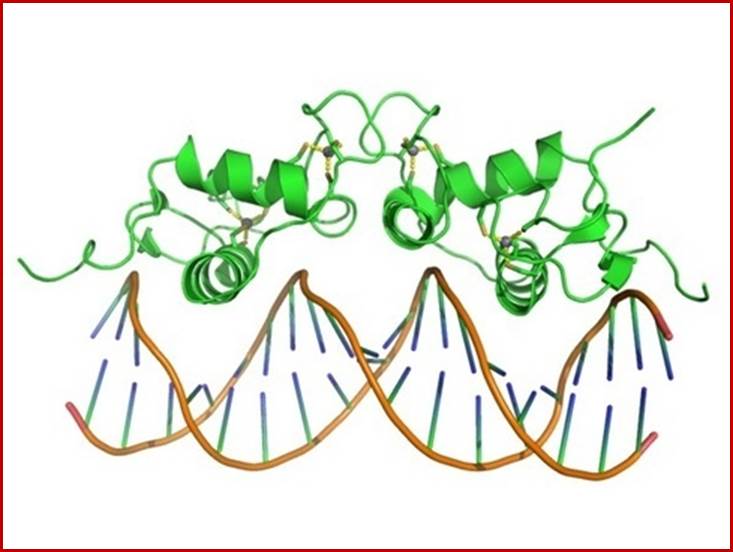
The Zn finger domain fits nicely in to major grooves defined by their DNA sequence and also the sequence of protein amino acids; www.quora.com
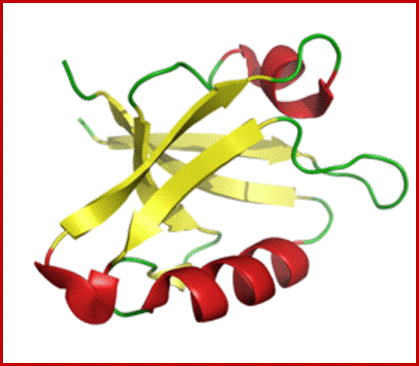
PDZ domain of the GOPC (Golgi-associated PDZ and coiled-coil motif-containing protein) protein.;
Domain: The concept of the domain was first proposed in 1973 by Wetlaufer after X-ray crystallographic studies of hen lysozyme and papain and by limited proteolysis studies of immunoglobulin’s. Wetlaufer defined domains as stable units of protein structure that could fold autonomously. In the past domains have been described as are compact structures, have function and evolve, help in folding. Domains are larger associations of two or more secondary structures, two or more super secondary elements, or mixtures of two or more secondary that are functional, they are called Domains. They can also be known as 'folds', and 'modules'. Each domain forms a compact three-dimensional structure and often can be independently folded and stable. Molecular evolution uses domains as building blocks and these may be recombined in different arrangements to create proteins with different functions. Domains are specially separated unit of the protein structure; they may have sequence and or structural resemblance to another protein structure or domain or it may have specific function associated with it.
Many proteins consist of several structural domains. One domain may appear in a variety of different proteins. As we will see later in Protein Tertiary Structure domains are independently folding units of tertiary structure and contain between 35 and 300 or more amino acid residues. Note that some other authors may present slightly different definitions of the terms above and we will look at some definitions later in Protein Tertiary Structure. The shortest domains such as zinc fingers are stabilized by metal ions or disulfide bridges. Domains often form functional units, such as the calcium-binding EF hand domain of calmodulin. Because they are independently stable, domains can be "swapped" by genetic engineering between one protein and another to make chimeric proteins.
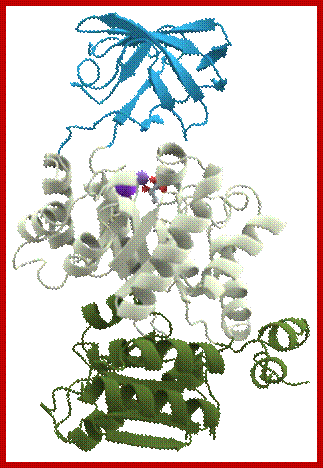
Pyruvate kinase with three domains; https://en.wikipedia.org/wiki/
Wikipedia; Wikipedia; http://www.thesgc.org/
Pyruvate kinase (see the figure above), a glycolytic enzyme that plays an important role in regulating the flux from fructose-1,6-biphosphate to pyruvate. It contains an all-β nucleotide binding domain (in blue), an α/β-substrate binding domain (in grey) and an α/β-regulatory domain (in olive green), connected by several polypeptide linkers. Each domain in this protein occurs in diverse sets of protein families. There are now over 2,600 classes of protein domains reported on Pfam (see online links box) — a manually curated database of protein-domain families — that are encoded by genes in the human genome NIHNCBI. Of these, 221 domains in proteins are found that are encoded by cancer genes. There are 27 cancer genes encode protein-kinase domains.
Rosmann fold; http://www.proteinstructures.com/
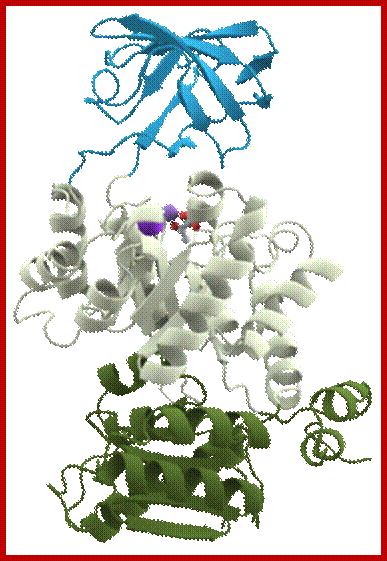
Pyruvate kinase, a protein with three domains; https://en.wikipedia.org
For more information on the number of domains in a protein and their folding, one needs to consult CATH or SCOP, two databases dedicated to fold classification. Although one needs to be aware that CATH and SCOP use slightly different terminology in domain assignment. According to SCOP, defined by scope, there are 1393 (1282) different folds. CATH comes from the first letters in Class-Architecture-Topology-Homologous superfamily. According to the latest release, there are currently 173536 domains in CATH (http://www.proteinstructures.com/).
For example Src3 (SH3) is a small domain consists of 50amino acid residues involved in protein to protein interactions. The SH3 domain has characteristic 3D structure. They occur in diverse range of proteins with different functions, ex. Phosphatidylinositol, phospholipase and Myosin’s.

SH3 domain; https://www.ebi.ac.uk
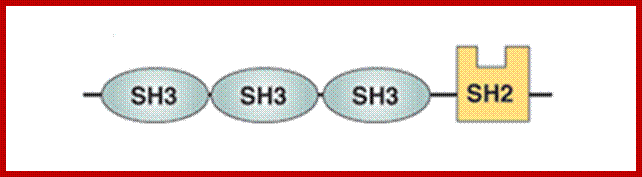
Domain N end contains three SH3 and SH2 domains, these are involved in protein-protein interactions; https://www.ebi.ac.uk.
DFU Domains: A large fraction of domains are of unknown function. A domain of unknown function (DUF) is a protein domain that has no characterized function. These families have been collected together in the Pfam database using the prefix DUF followed by a number, with examples being DUF2992 and DUF1220. There are now over 3,000 DUF families within the Pfam database representing over 20% of known families.
What are protein families: They are a group of proteins that share common evolutionary origin, have related functions and similarities in sequence and structure. They are arranged into hierarchies (closely related groups). Super superfamily the term indicates a large group of distantly related proteins and subfamily, a small group closely related. Below is the hypothetical figure showing hierarchy.
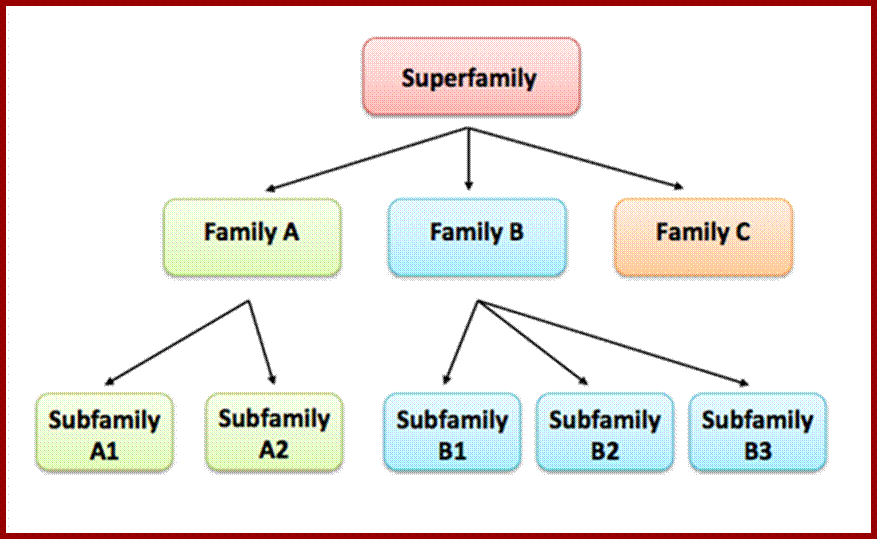
A hypothetical Protein family based on domain, showing relationship between super families, family and subfamily members. https://www.ebi.ac.uk/training/online/course
Rotational structural features of proteins; Prof. G. N. Ramachandran’s (IISc), a perspective;
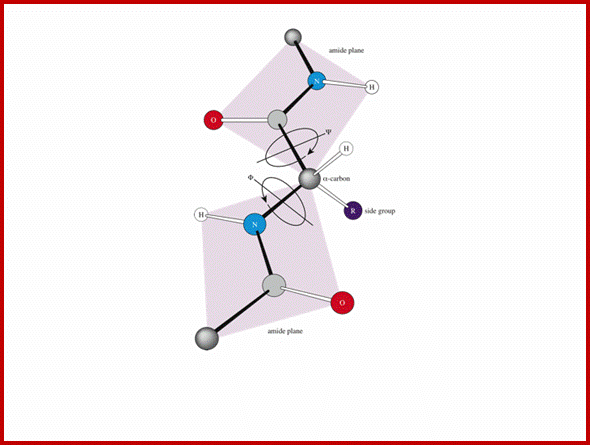
The backbone of a polypeptide chain is the C-N peptide bond between amino acid residues. Every polypeptide chain possesses one amino free end (N end) and the other and contains a free carboxyl group (C end). The peptide C-N region is planar and it is not free for rotation on its own axis. But the a carbon with its R group is pliable and it can freely rotate on its axis. Thus C-N planar group imposes significant constraints upon the shape of the polypeptide chain.
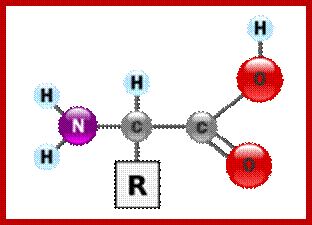
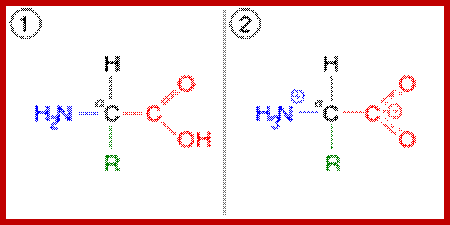
The generic structure of an alpha amino acid in its un-ionized form; An amino acid in its (1) un-ionized and (2) zwitterionic forms; https://en.wikipedia.org
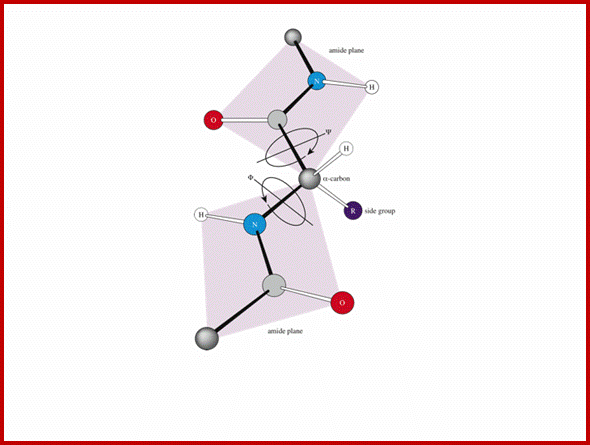
Peptide bond linking two amino acid residues, planar and rigid, but the alpha carbon is can go through rotation. biologicalphysics.iop.org


Authors of this website (grkraj.org) is my most favorite professor in IISc, when I invited him to give a lecture to BSc, students in National College, he came and gave the lecture. He was very happy to teach such students;
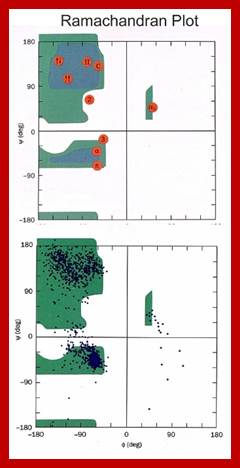
G.N Ramachandran’ plot.
Gopala Samudram Narayana Ramachandran, G.N. Ramachandran’ plot (also known as a Ramachandran diagram or a [φ, ψ] plot), originally developed in 1963 by G. N. Ramachandran, C. Ramakrishnan, and V. Sasisekharan, is a way to visualize backbone dihedral angles ψ against φ of amino acid residues in protein structure. The figure at left illustrates the definition of the φ and ψ backbone dihedral angles (called φ and φ' by Ramachandran). The ω angle at the peptide bond is normally 180°, since the partial-double-bond character keeps the peptide planar. The figure at top right shows the allowed φ, ψ backbone conformational regions from the Ramachandran et al. 1963 and 1968 hard-sphere calculations: full radius in solid outline, reduced radius in dashed, and relaxed tau (N-C alpha-C) angle in dotted lines. Because dihedral angle values are circular and 0° is the same as 360°, the edges of the Ramachandran plot "wrap" right-to-left and bottom-to-top. For instance, the small strip of allowed values along the lower-left edge of the plot is a continuation of the large, extended-chain region at upper left.
http://swissmodel.expasy.org/
In the diagram above the white areas correspond to conformations where atoms in the polypeptide come closer than the sum of their van der Waals radii. These regions are sterically disallowed for all amino acids except glycine which is unique in that it lacks a side chain. The red regions correspond to conformations where there are no steric clashes, i.e. these are the allowed regions namely the a-helical and a-sheet conformations. The yellow areas show the allowed regions if slightly shorter van der Waals radii are used in the calculation, i.e. the atoms are allowed to come a little closer together. This brings out an additional region which corresponds to the left-handed a-helix.
Another important factor that contributes to the orientation of the polypeptide chain in its stable form, in its least energy state, is R groups present at a carbon of each amino acid residue. They contribute to the configuration and conformation of the polypeptide chain in its entirety. Configuration denotes the arrangement of substituent groups in space and their position is always fixed.
The configuration cannot be changed without breaking one or more covalent bonds. On the other hand conformation refers to special arrangement of substituent groups where they are free to assume different positions without breaking any bonds. The side groups i.e. R groups of each amino acid residue strongly influence the conformation of the polypeptide chain and the possible conformations is infinite. In addition, the pH of the medium, thermal rotations, electronic energy, binding of other molecular adducts and such molecules have strong influence on the conformation.

Proteins can be crystallized; such crystals can be subjected to X-ray diffraction to understand the details of the Protein structure. www.swift.cmbi.ru.nl
Fundamental aspect of X-ray diffraction is the preparation of a very good crystal.
A good rule of thumb:
Poor crystals ==> poor, unpublishable results and low precision.
Growing crystals;
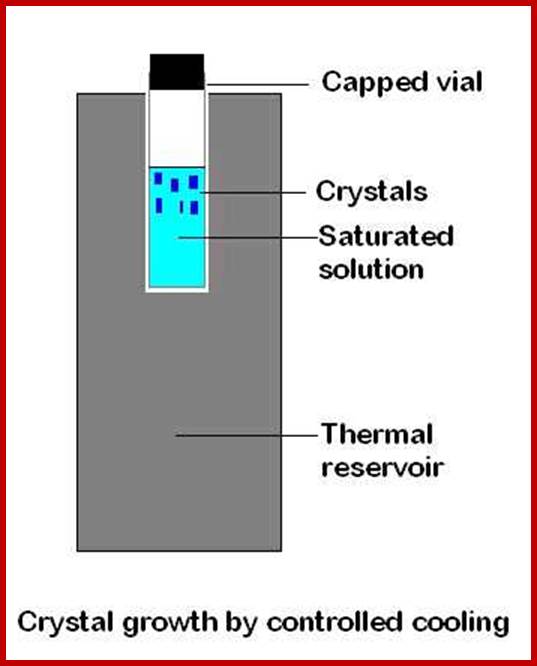
A simple model for growing a crystal; http://www.nottingham.ac.uk/
The key with each of the crystallisation methods is to allow the crystals to grow slowly. The vapour diffusion or solvent evaporation methods proceed too quickly if the flask containing the solution is left uncovered. Covering the flask with Parafilm and poking several small holes in the film with a needle is an excellent means of slowing down the vapour diffusion. http://www.chem.uzh.ch/; Crystals should be examined closely under a microscope, preferably one that has polarising capabilities.
For X-ray diffraction we must have a single crystal. The crystals should be transparent and appear to contain no flaws when viewed under the microscope. Crystals that are cloudy have cracks, appear to have other crystals buried inside or intergrown crystals protruding from the side should be rejected. Crystals that look like a bird's feather, a fern leaf, a dandelion seed or a star are not single crystals and are totally unsuitable. Sometimes very thin plate-like crystals or needle-like fibres stack themselves into what look like single crystals and are not immediately obvious to the inexperienced eye. Crystals that appear to have many parallel lines running along the length of the crystal may be suffering from this effect and should not be used. Ideally the best crystal is a sphere. This is to minimize absorption effects. The next best choice is a cube or a prismatic crystal with each dimension roughly equivalent.
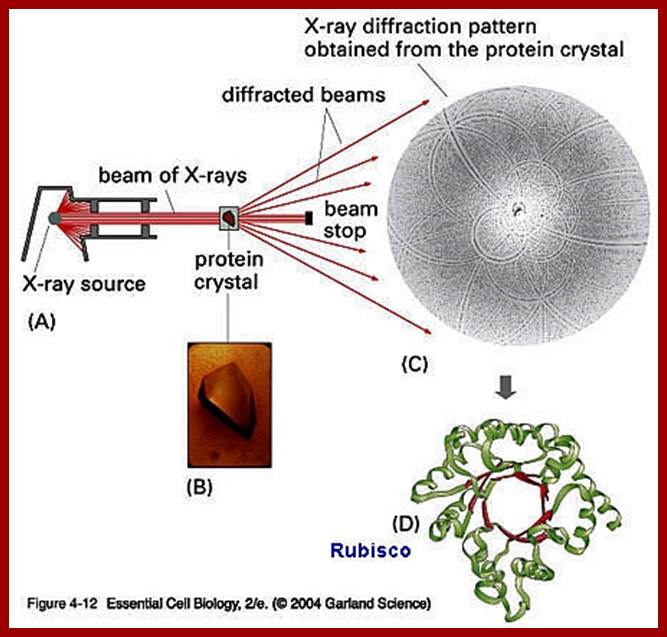
X-ray diffraction of Rubisco protein
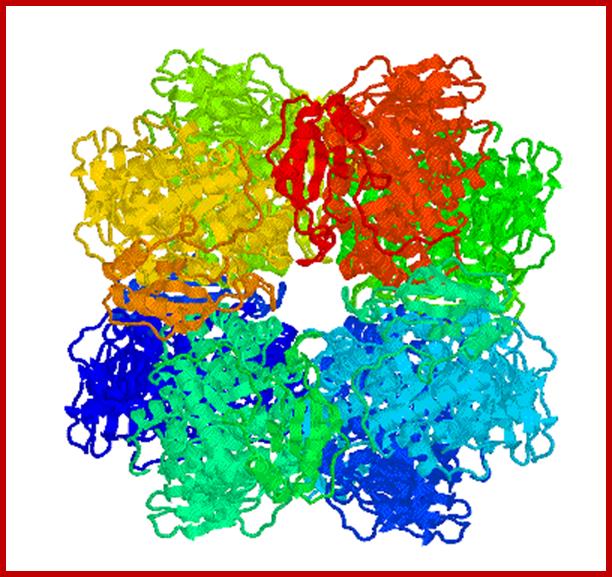
RubisCo octomer; http://www.medicaldaily.com/
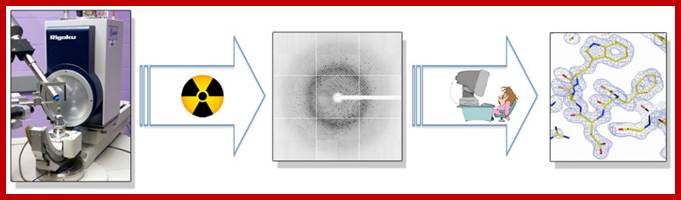
Above-X-ray diffraction of RubisCo protein crystal; http://www.lifesci.dundee.ac.uk/
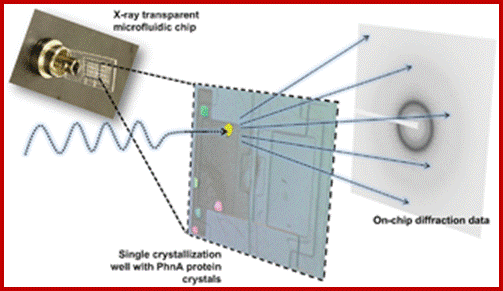
A microfluidic approach for protein structure determination at room temperature viaon-chip anomalous diffraction; An X-ray compatible microfluidic platform for de novo structure determination of proteins.; www.pubs.rsc.org
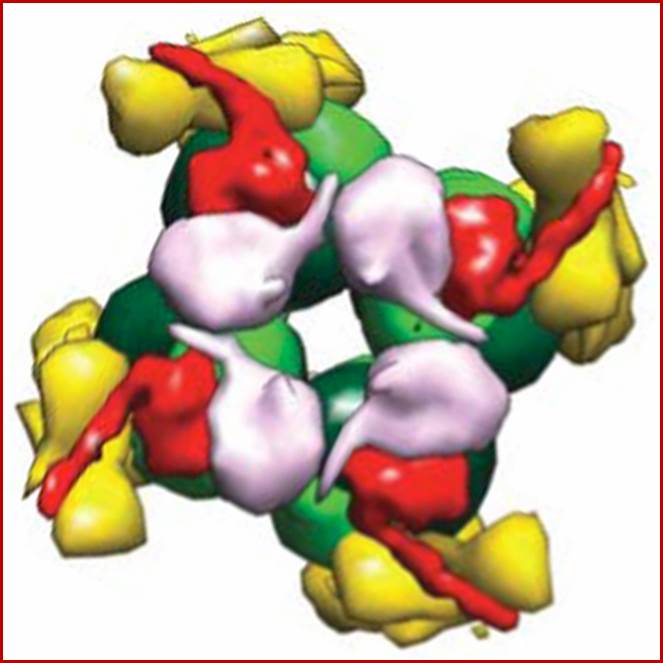
RuBisCo octamer; www.cipsm.de
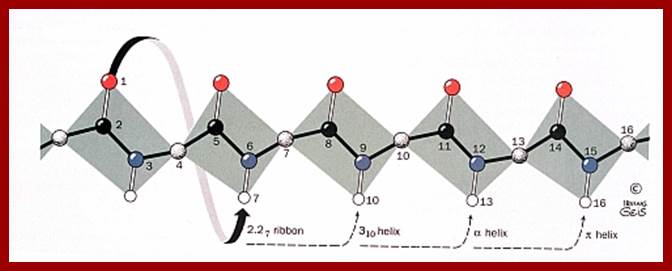
SECONDARY STRUCTURE:
Helical:
If a polypeptide chain is suspended in water, due to the interaction between forces that are operating on R groups and the forces exerted by dipolar water, the protein assumes
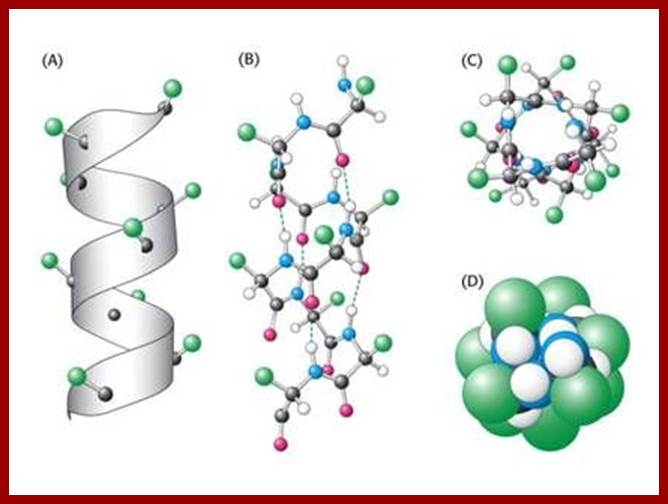
Helical form; which may be right handed helix a or left handed helix (b). ex., keratin proteins (hair proteins) assume right handed helix called a helix, in which for every 0.5 mm to 55 mm length the protein takes one complete turn consisting of +/- 3.6 amino acid residues.
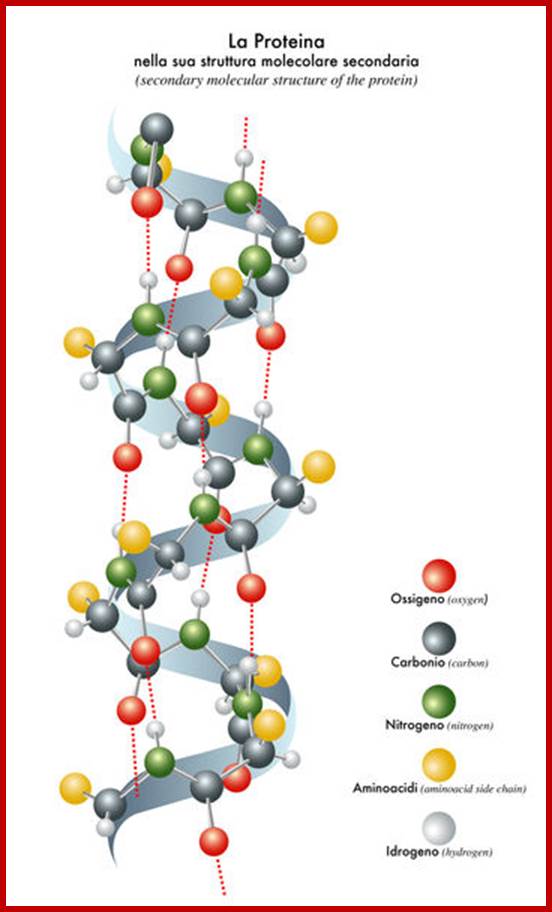
Secondary structure; http://www.biologyreference.com/
The R groups from each amino acid stick out of the helix. Because of this the inter chain hydrogen bond formation, between the electronegative nitrogen of one coils with the carbon oxygen of the third amino acid found in the next coil, is greatly facilitated. The hydrogen bondings are oriented parallel to the long axis of the helix. Most of the proteins exhibit right handed a helix.
![]()
It should also be noted that not all polypeptides should have helical conformation. For example polylysine polypeptides, at pH 7.0 do not assume any helical structure but remains as a random chain. Instead, the same polypeptide chain at pH 12 assumes a helical from. This is because at pH 7.0, the R group of all lysine residues having positive charges repels each other and prevents intra chain hydrogen bonding. On the contrary, at pH 12, the R groups are neutralized, and they do not exert any repelling forces, instead hydrogen bonding between intra chains is favored, hence they assume a helix. In the case of poly-isoleucine polypeptide chains, a helical conformation is prevented because of stearic hindrances exerted by bulky hydrophobic R groups.
The conformation in polyprolines is slightly different, because the N group is actually within the heterocyclic ring which forms the R group of the amino acid. This prevents the rotation on its axis which in turn prevents the hydrogen bonding within the polypeptide chain. If such proline residues are present with in a polypeptide chain, the helical confirmation at such regions is prevented and assumes B chains.
The above mentioned features strongly suggests that a polypeptide chain generally assumes a helical form, but it need not be throughout the length of the chain and a part of the chain may be in random form or it may assume any form which depends upon the amino acid sequences in that position. It also explains how the side groups exert influence in the 3-D conformation and stability of the protein chain.
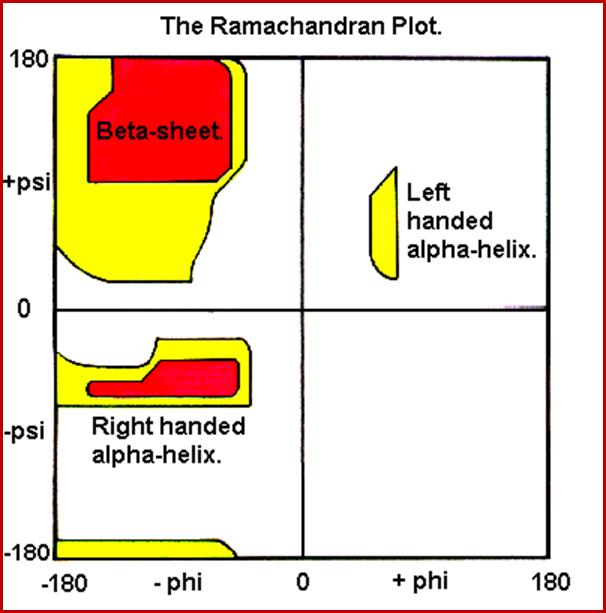
Prof. G.N. Ramachandran (late) from I.I.Sc, Bangalore has proposed another restraint with regard to the angle of rotation along the Ca -C1 axis. He has determined the most stable angles of rotation for the right handed helix between the C-R to C-0 = a = 47 degree and C-R to NH is Q=57 degree. The entire conformation of the protein chain falls within the allowed territory; called Ramachandran’s plot.
The two torsion angles of the polypeptide chain, also called Ramachandran angles (after the Indian physicist who first introduced the Ramachandran plot), describe the rotations of the polypeptide backbone around the bonds between N-Cα (called Phi, φ) and Cα-C (called Psi, ψ). The Ramachandran plot provides an easy way to view the distribution of torsion angles of a protein structure (RAMACHANDRAN GN, RAMAKRISHNAN C, SASISEKHARAN V., J Mol Biol., 7:95-99).
Definition:
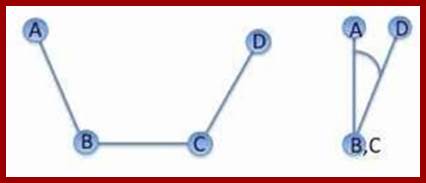
Torsion angles are dihedral angles, which are defined by 4 points in space. In
proteins the two torsion angles phi and psi describe the rotation of the
polypeptide chain around the two bonds on both sides of the Ca atom:
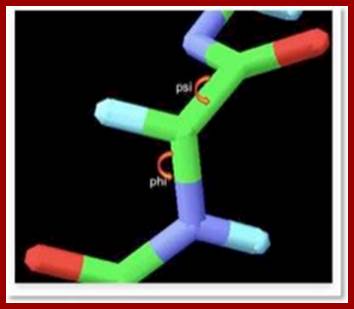
http://www.proteinstructures.com/
The Ramachandran angles in proteins are restricted to certain values, since some angles will result in steric clashes between main chain and side chain atoms in the polypeptide. In addition, for each type of the secondary structure elements there is a characteristic range of torsion angle values, which can clearly be seen on the Ramachandran plot: on the left plot the region marked alpha is for alpha-helices and beta is for beta-sheet. http://www.proteinstructures.com/
G.N Ramachandran's rotation along the C alpha-C1 axis in proteins; https://www.google.co.in
Beta chains:
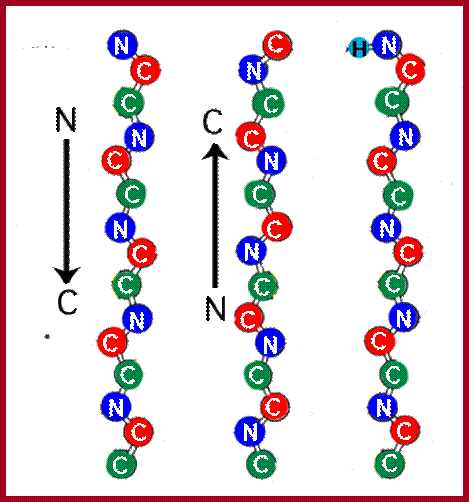
http://www.brooklyn.cuny.edu/
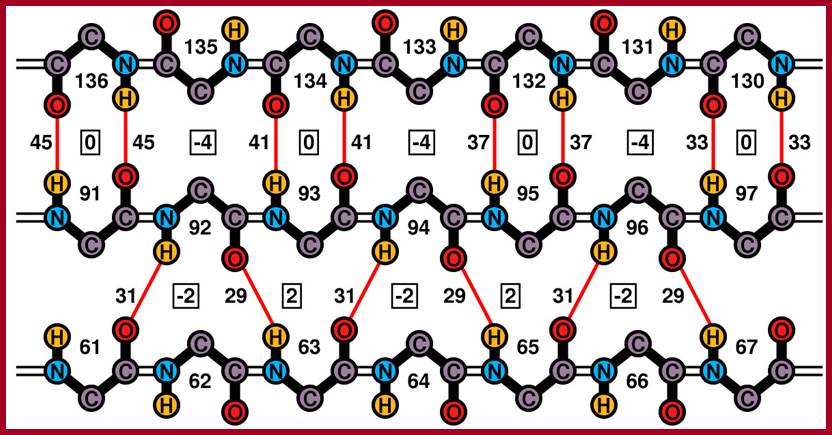
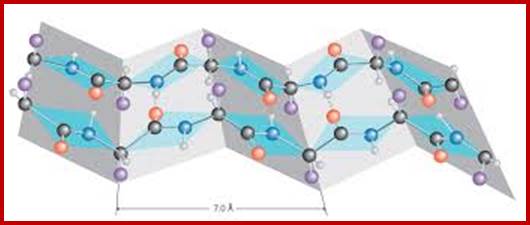
Ideal cases of a parallel and anti-parallel beta sheets; Residue numbers are surrounded by the backbone atoms of the respective residue, differences of hydrogen bonds are positioned next to the respective bonds and second differences are placed in boxes Zig-Zag model of Beta chains; http://www2.chemistry.msu.edu/ http://www.biomedcentral.com/
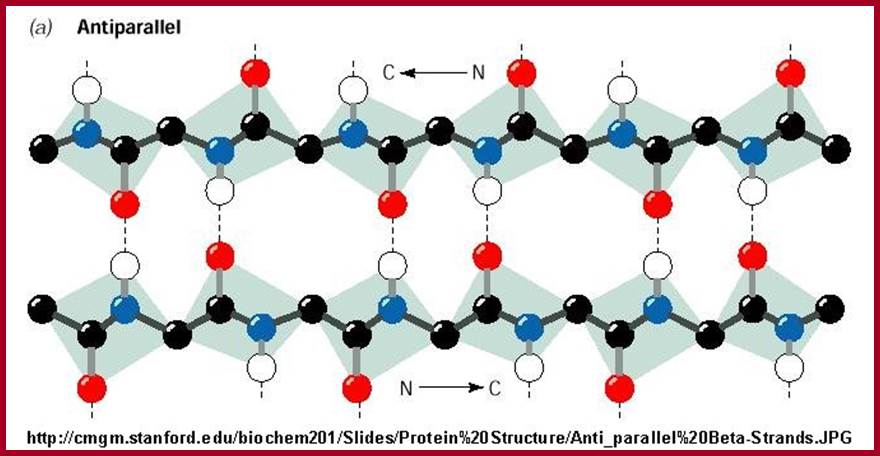
Antipararallel chains cross linked in Beta sheets. www.teaching.ncl.ac.uk
http://bioinfo.au-kbc.org.in/
Beta pleated sheets; www.drgpdreamdot.com
Primary helix or beta forms can lead to folding and compaction; In folding hydrophobic region close to each other cohese leaving hydrophilic a.a chains at the outer surface thus the protein assumes a 3-D structural fold.
Tertiary and Quaternary structures;
The helical polypeptides need not be a straight helical chain and it can further fold or associate with other R- groups of the same protein chain. They go through folding and the folds are stabilized by cross bonding between amino acids; www.imgarcade.com
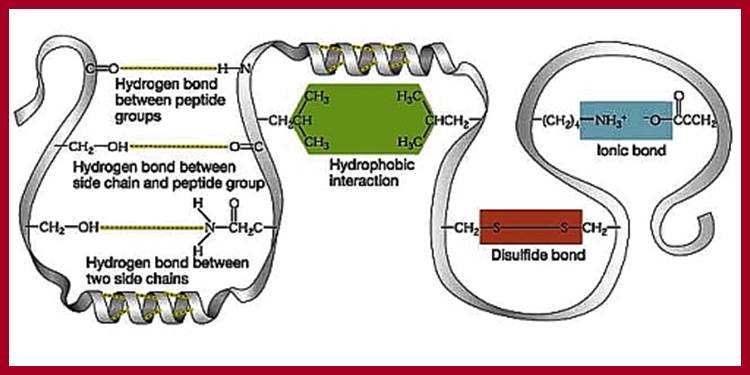
Four interactions stabilize the tertiary structure of a protein: (a) ionic bonding, (b) hydrogen bonding, (c) disulfide linkages, and (d) dispersion forces.; 2012books ; www...lardbucket.org
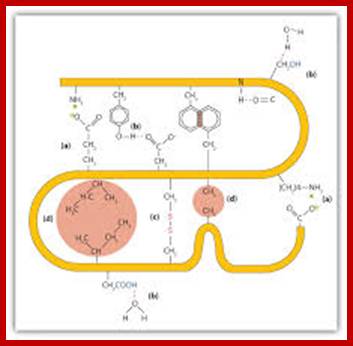
www.kimberlybiochemist.wordpress.com; www.alevelnotes.com
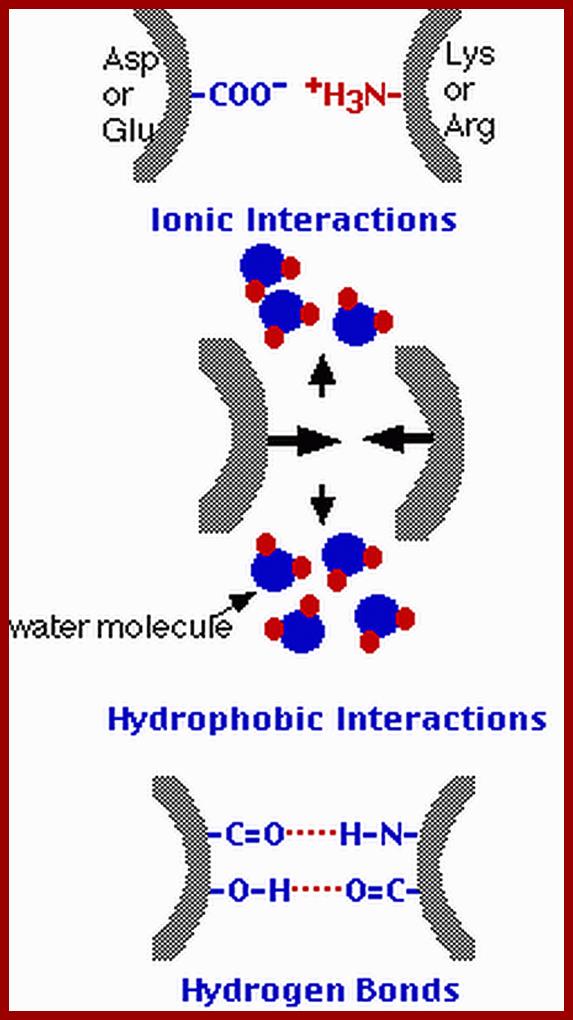
Ionic, hydrophobic and hydrogen bonding interactions between protein chains. http://users.rcn.com/kimball
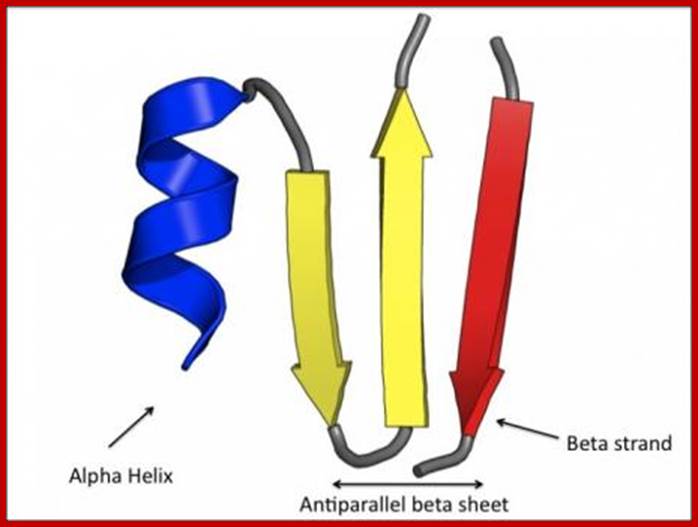
Proteins contain Combination of alpha helical and beta sheets; www.download.igb.uci.edu
![]()
This is the structure of triose phosphate isomerase, an enzyme which has a polypeptide consisting of alternating beta strands (yellow arrow symbols and alpha helix segments (red ribbon).
Turns are marked in blue, and loops
are random coils white;
Proteins like this fold as consecutive beta-alpha-beta units, which allows the beta strands to
line up into a parallel rather than antiparallel beta sheet.
The parallel beta sheet typically has non polar amino acids on both sides. If
all the alpha helices line up on one side of the beta sheet, the beta sheet
wraps around to form a parallel beta barrel with alpha helix on the outside.
Triose phosphate Isomerase; www.chembio.uoguelph.ca; Tertiary structural organization-
combination helical and beta sheets; www.chembio.uoguelph.ca
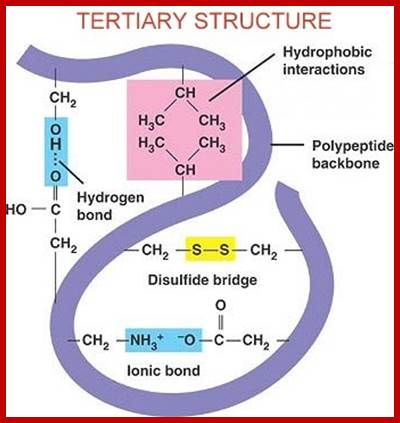
Proteins fold on their own and often aided by chaperones to produce a 3-D structure. molecular chaperones are proteins that assist the covalent folding or unfolding and the assembly or disassembly of other macromolecular structures. This happens in the lumen of endoplasmic reticulum and also outside ER. Specific bonds form at specific positions and these bonds provide stability to protein’s 3-D structures. Such structural form is called tertiary structure. The R groups that are responsible for such interaction may bring about hydrogen bonding, ionic interactions, hydrophobic interaction, and covalent cross bridges like sulfhydril bonds. Among the above mentioned attractive forces, sulfhydril bonding in the strongest. Such covalent S-S bonds within the chain bring about the folding of the chain into specific 3-D conformation. Such 3-D conformation is very much essential for specific structural organization and functions.
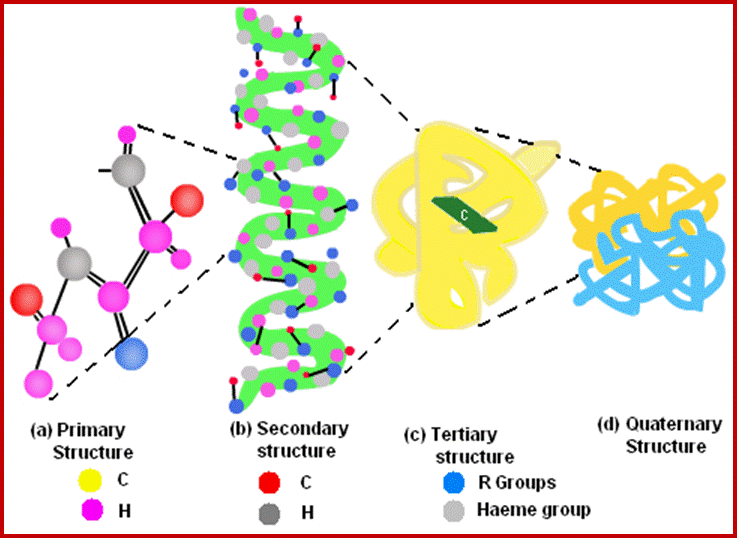
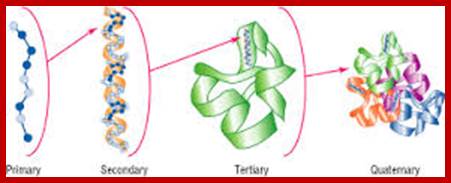
The four levels of protein structure. The sequence of amino acids, represented by blue dots, joined by peptide bonds, comprise the primary structure. The properties of the constituent amino acids, in the context of the cellular environment, largely determine spontaneous formation of the higher-level structure that is essential for protein function.; http://www.piercenet.com/
QUATERNARY STRUCTURES
A protein may be made up of one polypeptide chain or many polypeptide chains (subunits), the former is called monomer protein and the latter is referred to as polymer or oligomeric proteins.
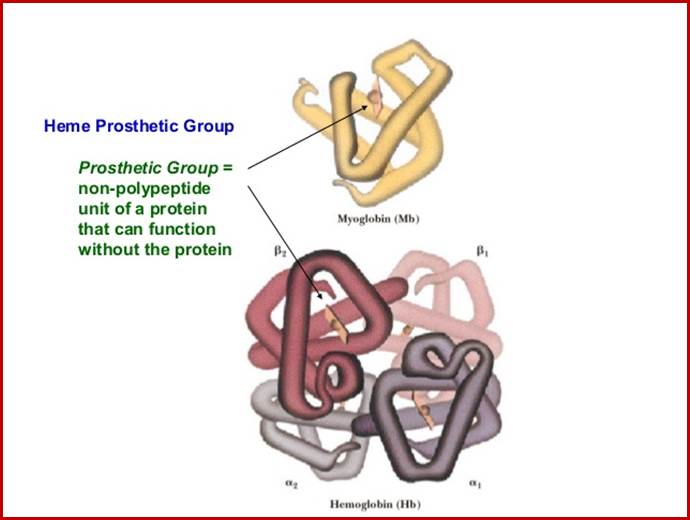
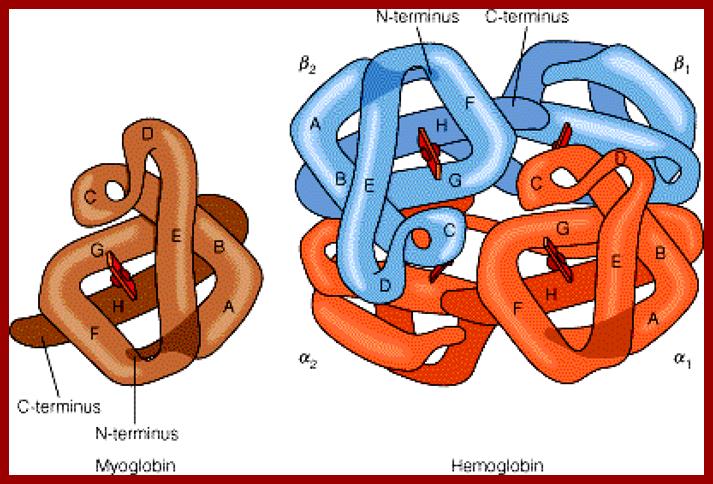
Myoglobin monomer and hemoglobin polymer of Hemoglobin protein; ufq.unq.edu.ar
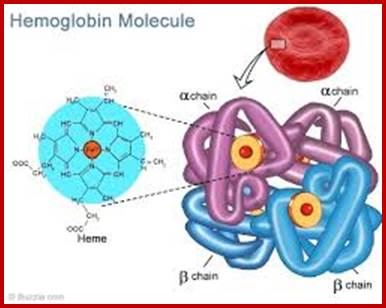
Hemoglobin four subunits called globin subunits and in the centre it has a heme group; www.buzzle.com

http://science.halleyhosting.com/
http://cbm.msoe.edu/
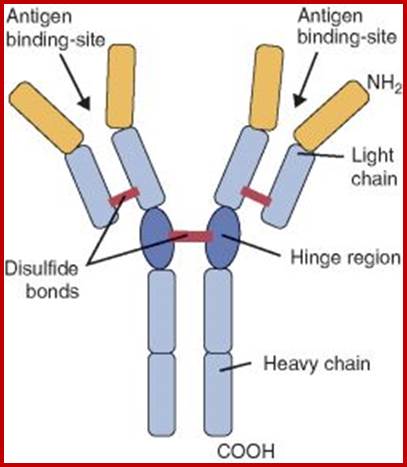
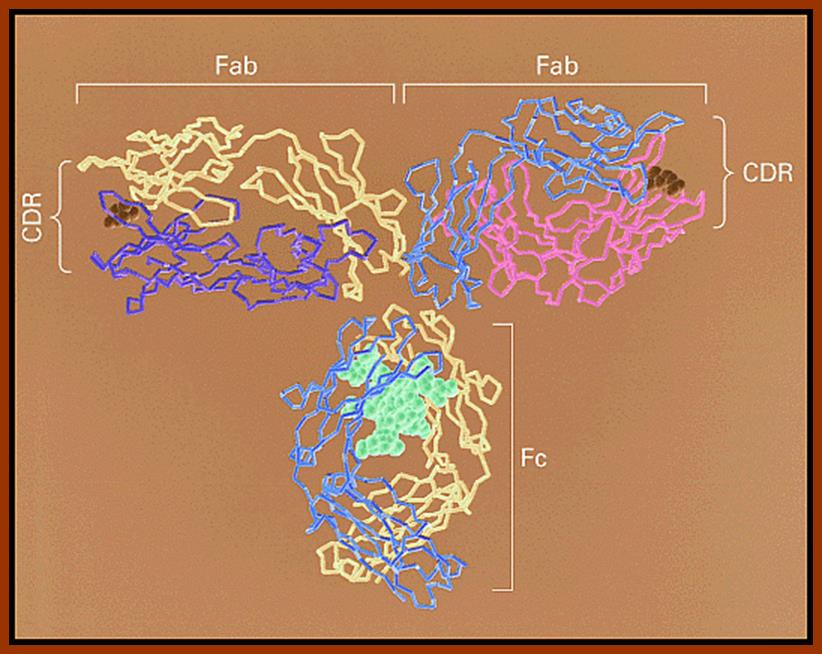
Immunoglobulins; www.uvm.edu
The dimer or polymer proteins may be made up of similar polypeptides or different polypeptides, so they are called homopolymers and heteropolymers. Such an association of more than one protein is referred to as quaternary structures. The binding forces responsible for such an association may be due to metal ions, hydrophobic interactions or ionic interactions.
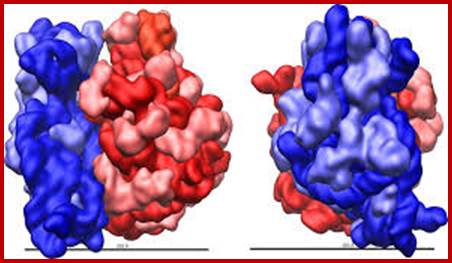
Ribosome is made up of 70 or more proteins associated with rRNA in 3-D; www.homes.cs.washington.edu
http://cbm.msoe.edu/
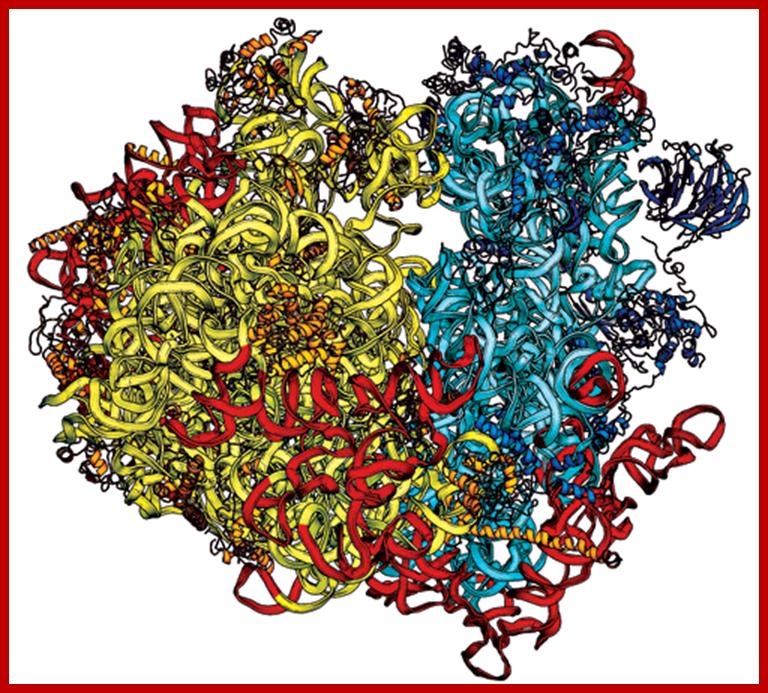
Yeast Ribosome structure; Jyllian N. Kemsley; https://pubs.acs.org/
Some simple combinations of secondary structural elements have been found to frequently occur in protein structure and they are referred to as supersecondary structure or motifs ex. Helix loop Helix, or beta-alpha beta, they are motifs..
[Several motifs join to generate a domain. The concept of the domain was first proposed in 1973 by Wetlaufer after X-ray crystallographic studies of hen lysozyme A sequence in the protein is folded into a structural feature that acts as a domain, the size of a domain can be 25 a.a to 250 or more. Zinc-finger domain, EF domain of Calmodulin is stabilized by S-S bonds. Each of the domains has independent function(s). ex. Alpha helix, bundle of four helix, a globin fold; Parallel beta-sheets-alpha-beta barrel-Triose phosphate Isomerase; Antiparallel sheets- immunoglobulins. At gene level exons of a mRNA codes for a sequence of amino acids can generate either a motif or a domain depending upon the length of the exon sequence. Many proteins do contain similar motifs and similar domains. The kind of domains and the number of domain’s structure and function is determined by the combination of exons. Assume in a given genome there are twenty thousand protein coding genes. Each produces an mRNA consisting of 20 exons; now one can calculate the number of similar different domains by permutation- combinations.
Motifs have structural and functional; a simple motif can be helix lop helix; two alpha helixes are joined by a simple loop ex. DNA binding motif and calcium binding motif.
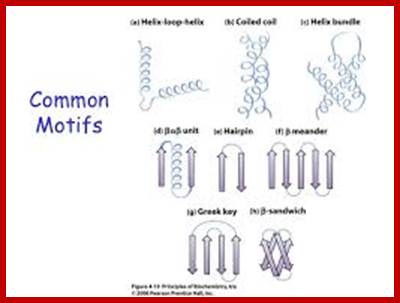
www.slideshare.net
CH domain; The CH domain consists four main α-helices connected by either long loop regions or two to three shorter helices. Three dominant helices form a parallel bundle against which the N-terminal helix packs at a right angle. Two CH domains in tandem pack such that the C-terminal helix of the first domain connects the two domains to create a single compact fold. - See more at: http://www.cellsignal.com/common/content/content.jsp?id=domains-ch#sthash.2zVrL9BU.dpuf; http://www.cellsignal.com/
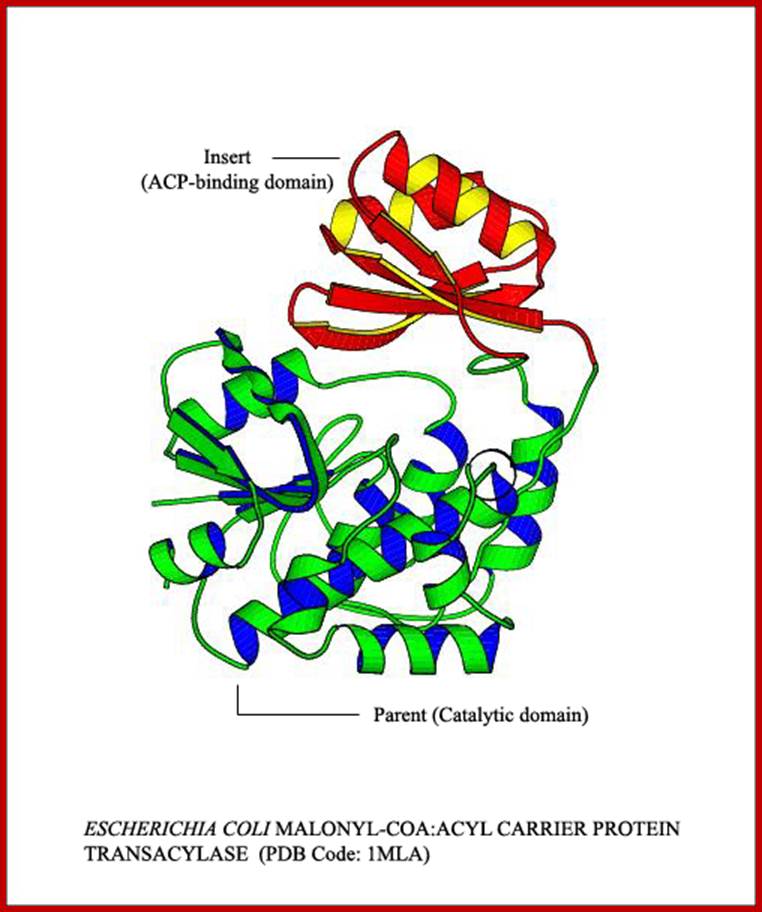
http://www.domins.org/faq.html
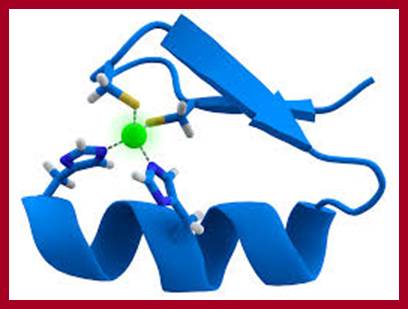
Zinc finger domain; pfam.xfam.org
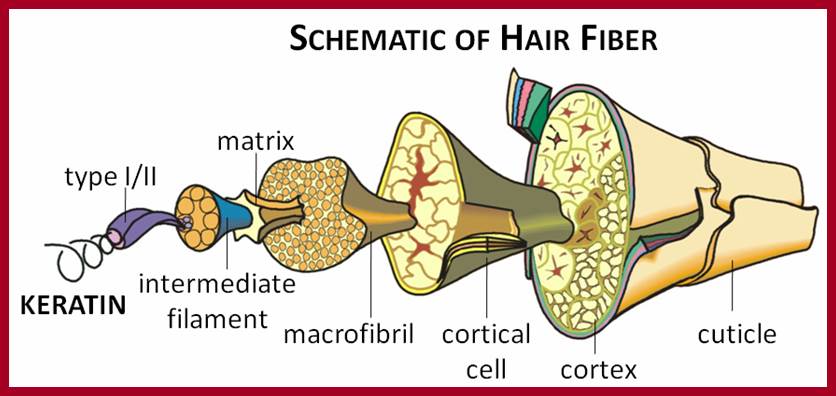
Human Hair fiber; www.hairkeratins.com
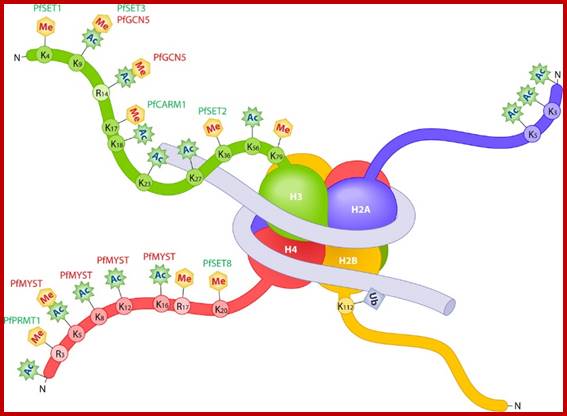
Histone octamers wrapped around by dsDNA; www.ec.asm.org
Protein-protein interactive domains
Shapes:
Globular proteins:
Globular proteins polypeptide chains which are folded into 3-D spherical structures are called globular proteins. Nearly 2000 and odd enzymatic proteins so far known are globular proteins. Such proteins are soluble in water and some are buried in the lipid core of the membranes; which depends upon the kind of peripheral amino acid residues found on proteins, ex. Hemoglobins, myoglobins, serum proteins are water soluble; Cytochrome oxidase, protein secretion proteins, etc. are lipid soluble proteins. The globular proteins contain specific areas or sites at which they bind to specific substrates for enzymatic reactions. The intergrity of the 3-D shape is essential for their normal functions.
FIBROUS PROTEINS
Many globular proteins organize into fibrous proteins. For example, a and B subunits of tubulins polymerize to form tubular microtubules. Similarly the G action units polymerize into functional filament called F-actins. Such proteins are often called pseudo fibrous proteins. But polypeptide chains of a keratin and B collagen proteins are considered as true fibrous proteins
Human hairs are primarily made up of numerous a keratin helical polypeptides. Such helixes are intertwined with each other by disulphide bonds between helical chains. All the chains in such structural fibers have NH2 or Carboxyl groups at the same ends. Basically, three such a keratin helical polypeptides are coiled to each other into a rope like structures called protofibrils. Even such protofibrils associate to form a micro fibril. Many such micro fibrils join together into a macrofibril. A large number of such macrofibrils join together to form a super coiled structure called hair.
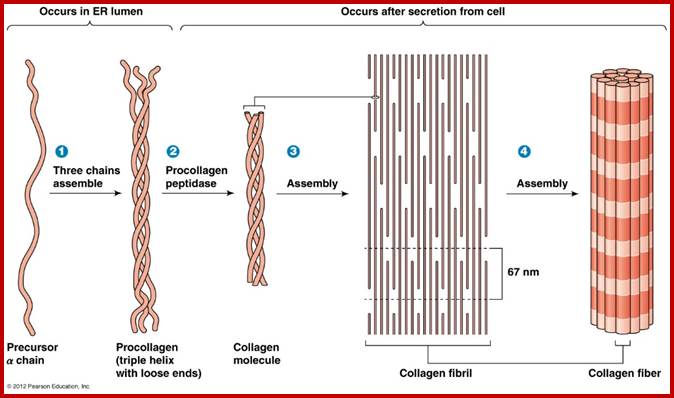
Collagens, the most abundant proteins in the ECM (25-30% of total protein in vertebrates), are secreted by cells, such as fibroblasts. Collagens are responsible for the strength of the ECM and form high tensile strength fibers and are prominent in tendons and ligaments. Collagens occur in a triple helix of three polypeptide chains and are high in glycine, hydroxy-lysine and Hydroxy proline. Collagen fibers are bundles of collagen fibrils which are, in turn, bundles of collagen molecules which consist of three alpha chains of collagen polypeptides. Procollagen forms many types of tissue-specific collagens.
Similarly collagen is another super coiled muscle fibrous protein found in the tissues of higher vertebrates. The basic unit of collagen fibers is tropocollagen which is made up of three helical polypeptides twisted to each other. Each tropocollagen is 300 mm long and 1.5 mm thick. Many such tropocollagen fibers are longitudinally arranged head to head to form long thick fibers. Many such fibers are longitudinally oriented to form muscle fibers called collagen fibers.
Silk fibrions or fibrions secreted by silk moths, spiders and other insects are insoluble proteins but they are supple and flexible in nature. Such fibers are different from a keratin fibers in their structure and flexibility. Keratins can be stretched to greater lengths, but silk fibrins cannot be stretched.
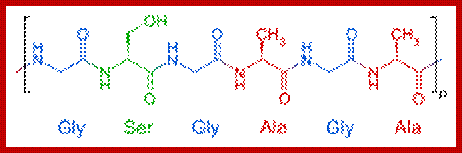
Bombax mori fibroin-(Gly-ser-gly-ala-gly-Ala)n; en.wikipedia.org
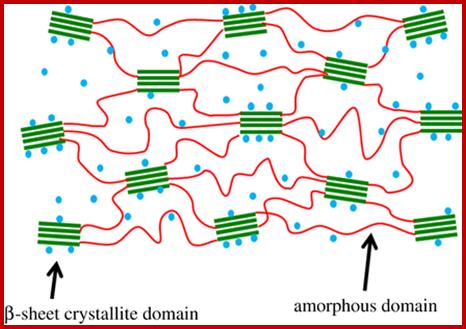
The illustration of β-sheet crystallite embedded in the amorphous matrix. Water molecules are represented by blue dots.;http://rsif.royalsocietypublishing.org/
Silk fibroins are actually made up of polypeptide chains with beta confirmation, wherein the chain is extended into zig-zag rather than helical conformations. Such zig zag fibers are oriented side by side parallel to each other and they are held to each other by interchain hydrogen bonds (See Figure). That is the reason why silk fibers look like stretched pleated sheet structures. The most significant feature of these proteins is the total absence of intra chain sulfhydril bonds and most of polypeptide chains are arranged parallel to each other.
FIBRO-GLOBULAR PROTEINS
Most of the cellular proteins fall into either into globular or fibrous types but some of the proteins found in both the animals and the plants (rarely), contain another class of proteins which have both the features. Such proteins are called fibro globular proteins.
www.quizlet.com
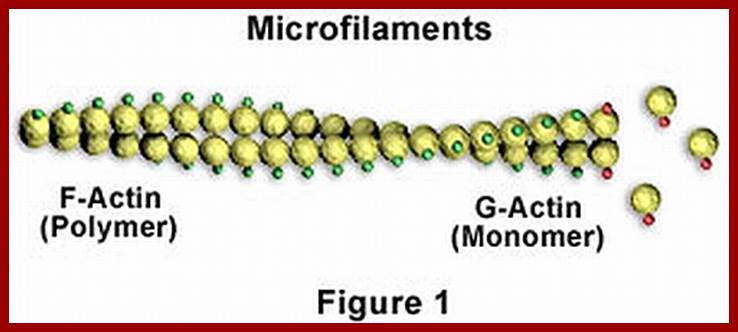
Microfilaments; www.studyblue.com
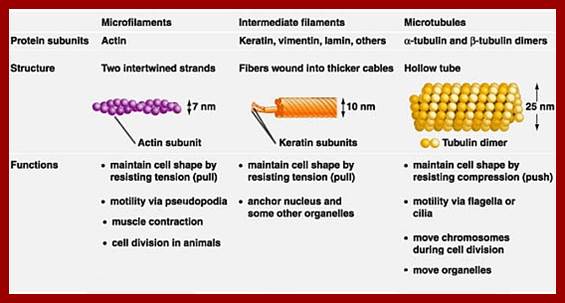
Actin, Keratin and Microtubules; www.pixgood.com
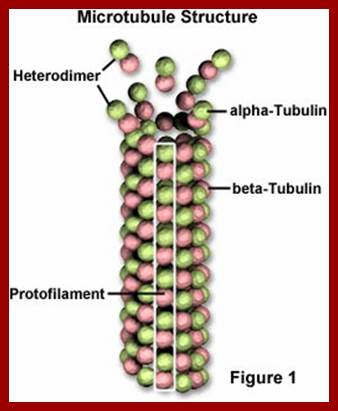
Microtubules: www.biology.ualberta.ca
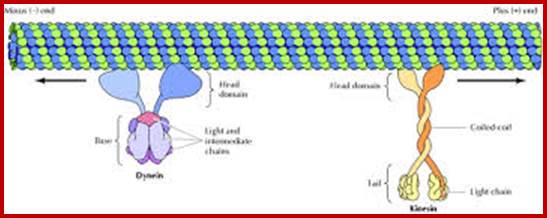
Microtubules with motor proteins; www.education-portal.com
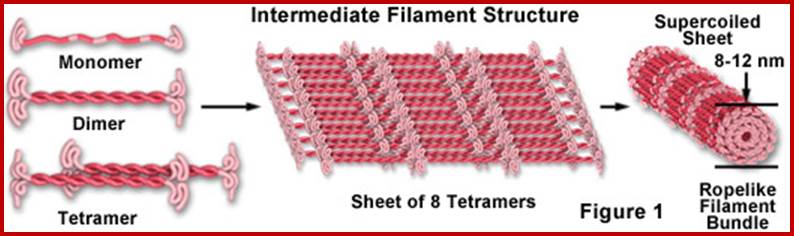
Intermediate filaments; www.micro.magnet.fsu.edu
The best example to illustrate such structural combination is myosin. It is made up of a head and a long tail. The tail consists of two long intertwined a helical polypeptides. On the contrary, the head consists of four polypeptide chains folded into globular structure; of which two are in continuity with tail fibers. The head proteins exhibit ATPase activity. The head and the tail protein of the myosin fibro globular proteins are interconnected by a region called hinge which exhibits random arrangement of the polypeptide chain.
Largest Protein known today is: TITIN:
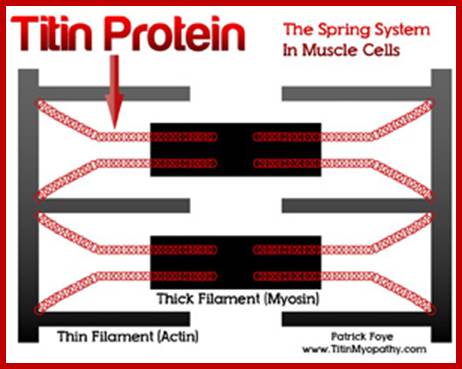
wwwcentronuclear.org.uk
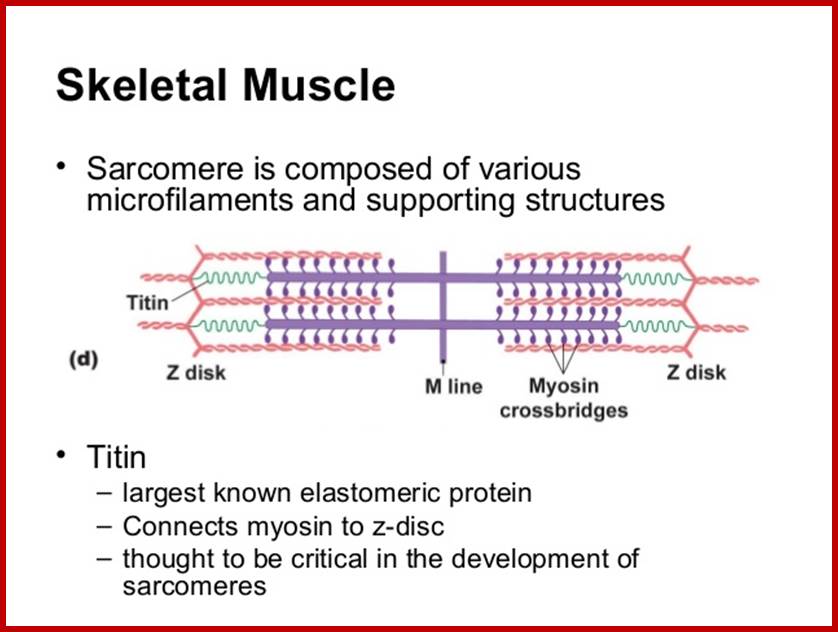
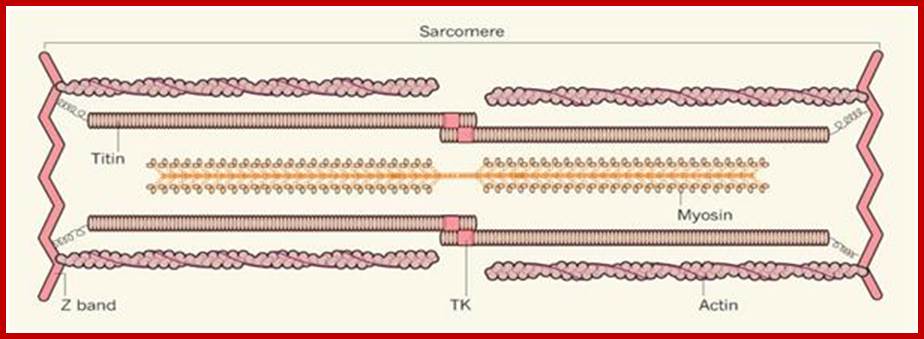
www.biol10202012-1blogspot.com
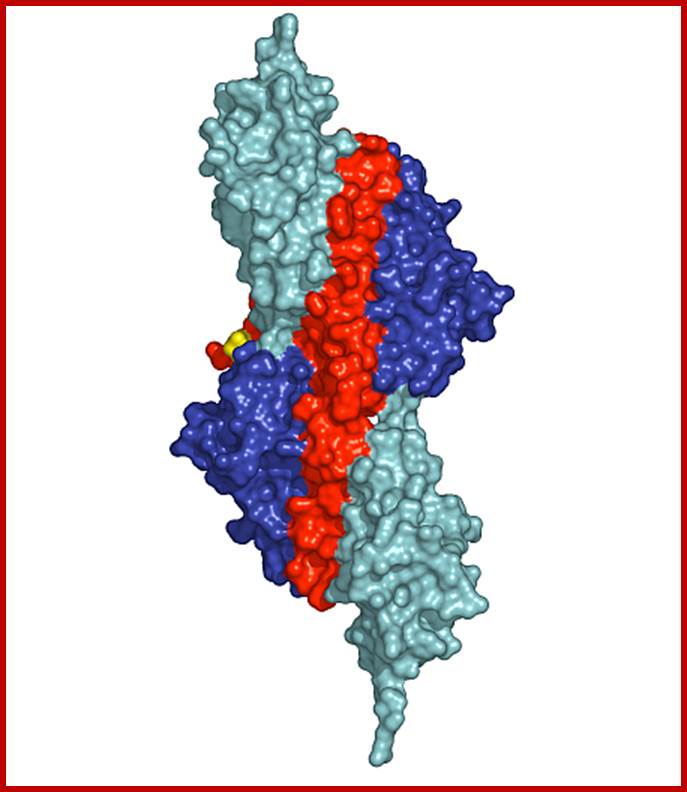
Surface presentation of the structure of the palindromic titin/telethonin/titin complex; Colour codes: titin IG-like domains, blue and cyan; telethonin, red and green. http://photon-science.desy.de/
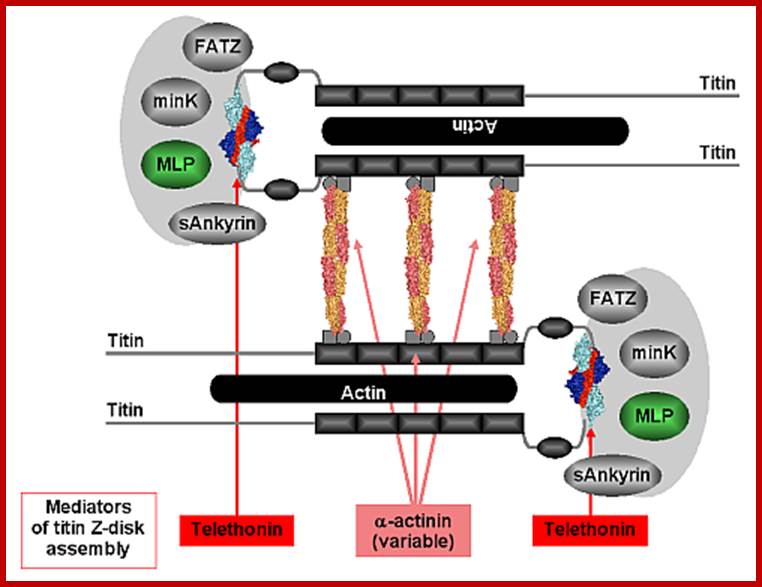
Figure 2: Model outlining the involvement of the titin/telethonin complex in the architecture of the sarcomeric Z-disk. Titin filaments are assembled by a dual Z-disk bridging system, by α-actinin rods on a variable number of titin Z-repeats (three bridges are shown), and by telethonin via the N-terminal IG domains Z1 and Z2. The titin N-terminus/telethonin complex forms a core that interacts with several ligands both inside and outside the sarcomeric Z-disk. http://photon-science.desy.de/
Protein functions:
The chemical composition and the structure of proteins are so diverse, their functions also vary. They are extra ordinary biomolecules endowed with a potentiality to provide structural stability to cell, determine the shape and perform myriads of functions. Though DNA act as the genetic material with all the information encoded within it, without functional protein, it is like a dummy tape without an instrument to play. No cellular component can be synthesized or processed without proteins. Their functions are pervasive; all biological activities are the functions of proteins. However there are exceptions to this rule. There are many RNA molecules in their specific structural form can perform enzymatic reactions, where they can cleave a bond and make a bond. Based on protein structure and functions a simple classification has been given below.
Enzyme proteins:
The most specialized proteins are enzymes and they act as biological catalysts. So far 10000 or more enzymes have been identified from bacteria, fungi, animals and plants. They are mainly responsible for biochemical activities of the cell.

RuBisco protein complex-Octamer; www.howplantswork.com
STORAGE PROTEINS:
Seed proteins like zein, gliedin etc are called storage proteins. They provide nutritional requirement of essential amino acids for animals. Such proteins are also stored in the white of eggs. E.g. Casein in milk and Ferritin in animal tissues.
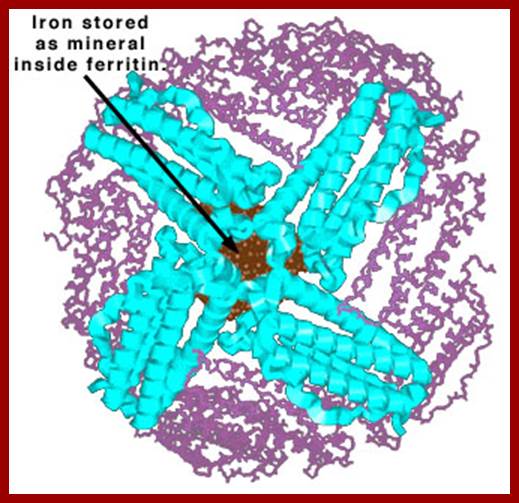 Ferritin iron sequestering protein; www.chemistry.wustl.edu
Ferritin iron sequestering protein; www.chemistry.wustl.edu
STRUCTURAL PROTEINS:
Proteins found in membranes, cytoskeletal fabric, capsids, collagen, elastin of muscles, mucoproteins of synovial fluids, keratin of hairs, nails, hoofs, borns, silk fibrions, spider webs, etc. are all structural proteins. They provide mechanical support and strength to various structural components of the cellular tissues.
TRANSPORT PROTEINS:
Certain cellular components are transported from one region of the cell to the other or from one region of the body to the other. Specific proteins are responsible for the transportation of various cellular components. Such proteins are called transport proteins. Microtubules and microtrabaculae transport sucrose in sieve tubes, microtubules and action filaments are involved in protoplasmic streaming. Hemoglobin, hemoeyanin and myoglobin transport oxygen in the blood of vertebrates and invertebrates. Serum albumin and B lipoproteins transport fatty acid components in the blood. Iron binding proteins and ceruloplasmins transport iron and copper respectively.
CONTRACTILE PROTEINS:
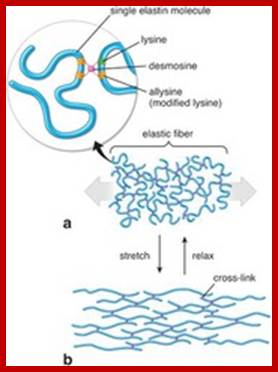
Elastin and fibrillins; www.quizlet.com
Flagillar proteins help in the movement of cells from one place to another. Cytoskeletal fabric is responsible for the protoplasmic movement. Muscular proteins control mechanical movement of organs and the body. Most of the above said movements are due to the activity of contractile proteins like action and myosin.
DNA and RNA binding Proteins;
A large number of proteins bind to DNA and RNA where DNA’s and RNA’s functions are executed. The DNA has encoded genetic information; to transfer the information proteins are required.
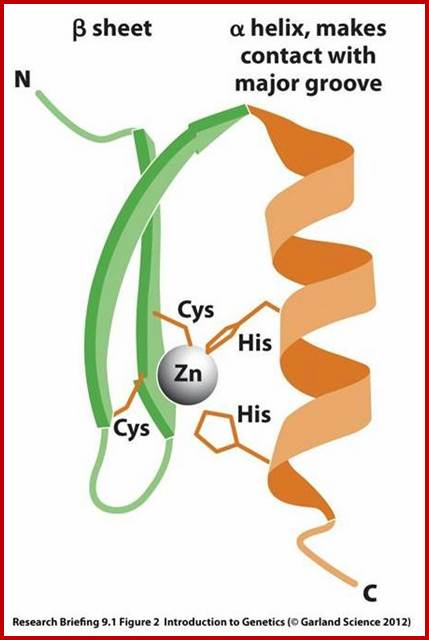
The motif facilitates binding of beta-sheets and alpha-helices; the latter can bind major grooves in DNA; Zinc finger DNA binding motif; www.mun.ca
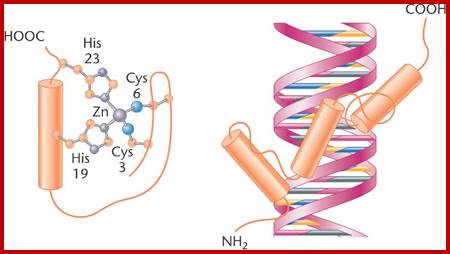
Zif Protein;Another motif comprising the DNA-binding domain of activator proteins is zinc finger, which contains cysteine and histidine amino acids bound to zinc atoms;www.bio3400.nicerweb.com

Helix lop Helix DNA binding protein; www.biologyreference.com
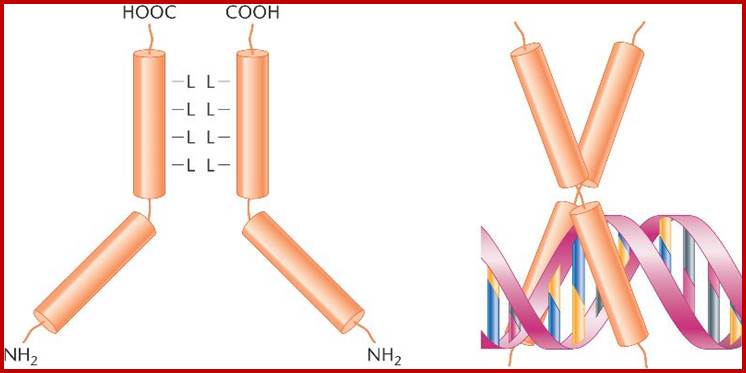
Leucine zipper is a protein dimer formed between leucine residues in parallel oriented alpha helices, bound to DNA; www.biochem.umd.edu
DEFENSE PROTEINS:
Antibodies are a class of serum proteins which act against the invasion of pathogenic bacteria, viruses or any other foreign substances. They are highly specific to the antigens. Interferons induce antiviral proteins against the attack of certain viruses in mammalian dells. Thus such proteins provide defensive mechanism against disease causing foreign agents. Fibrinogen and thrombin are another set of proteins, which prevent hemorrhage by blood clotting.
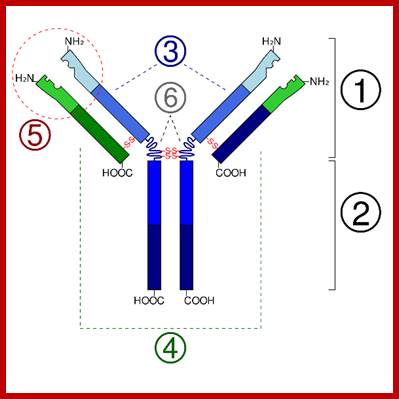
www..en.wikipedia.org
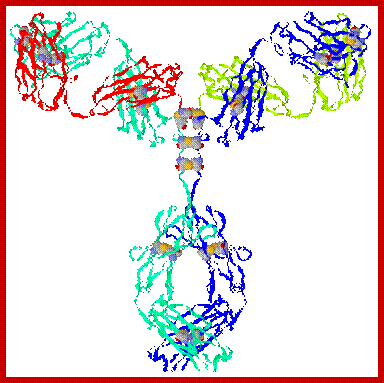
Immunoglobulin proteins-general; http://www.my-immunity.com/
TOXINS:
Certain pathogenic bacteria after infection release toxins which results in dehydration or food poisoning. Snake venom is another class of proteins which can cause death in man. Ricin produced by castor seeds and gossip in from cotton can easily kills persons. Thus proteins not only act as saviors as in the case of antibodies, they can also act as killers.
Diphtheria toxin; ;http://www.rcsb.org/
REGULATORY PROTEINS:
Animals produce certain proteins which control physiological activities, e.g. hormonal proteins. They are synthesized and secreted by endocrinal glands. Insulin controls the blood sugar level; growth hormone controls the growth of the body. Thus many such hormones play important roles in the life cycle of animals. Further more some of the proteins control transcription and translation, thus they control gone expression and differentiation. Even such proteins are called regulatory proteins.
GAL-4 protein; www.wikipedia.org
DENATURATION AND RENATURATION:
When proteins are heated or dissolved in certain solvents, their 3D structure, denatures and they become linear. This phenomenon is called denaturation; as a result, proteins lose their ability to perform their specific functions. On the other hand, the temperature of the said solution is brought back to the normal temperature, the sulfhydryl bonds are reformed and the 3-D shape is restored, so also its function. Such a process is called renaturation. On the contrary, if the solution is heated to boiling temperature, proteins undergo irreversible destruction.
When a solution containing proteins in their 3-D state are heated, to 40-50 degree C, the sulfhydryl bonds break and polypeptides open out into straight helical structure; this is called denaturation. And when the temperature brought to normal they fold back, it is called renaturation.
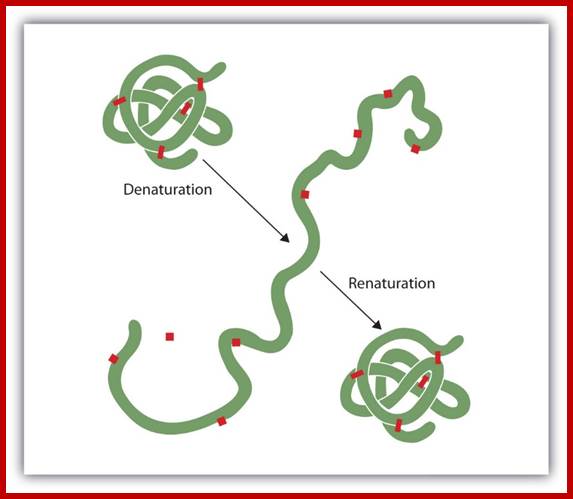
The denaturation (unfolding) and renaturation (refolding) of a protein is depicted. The red boxes represent stabilizing interactions, such as disulfide linkages, hydrogen bonding, and/or ionic bonds; .www. http://2012books.lardbucket.org/
Chemical bonds responsible for 3-D conformation, thus they bring about denaturation. Even changes in the pH of a solution bring about denaturation. Thus, it is clear, that the intracellular environment plays an Sodium dodisulphate, methyl mercury, mercury, etc. bind to proteins and break the important role in the function of proteins.
Molecular weight of few proteins:
The molecular weight of a protein depends upon the number and the kind of amino acid residues present in the chain. Added to this the number of sub-units present in a particular protein has to be taken into consideration for determining molecular weight of a protein complex. The molecular weight of individual polypeptides or the total mol. wt. of polymer proteins can be determined either by ultracentrifugation method or by column chromatography or electrophoretic methods. Using these methods the mol. wt. of and the number of sub-units present in various proteins have been determined. (See Table; only few proteins’ mol. wt is given).
|
Name of the protein |
Mol. Wt. In Daltons |
Number of sub-units |
|
|
Insulin |
5700 |
2 |
|
|
Ribonuolease |
12600 |
1 |
|
|
Lysozyme |
13900 |
1 |
|
|
Myoglobin |
16900 |
1 |
|
|
Hemoglobin |
64500 |
4 |
|
|
Aspirate transcarboxylase |
310000 |
12 |
|
|
Glutamine synthetase |
542000 |
12 |
|
|
Pyruvate dehydrogenase |
700000 |
160 |
|
|
TMV
|
40000000 |
2130 |
|
|
Titin |
27000-34350a.a; 3,816,030 mol.wt |
largest protein-monomer |
|
|
|
|
|
|

Titin is one of the largest proteins known as Titin (connectin), connects Z line to M line in sarcomeres; discovered by Reiji Natori in 1954, Kuan Wang and coworkers coined the term Titin; www.faculty.washington.edu
ENZYMES
In olden days wine-making was an art. They used to prepare wine from different sources like fruits, barley and wheat by subjecting them to a process called fermentation. But they did not know what the actual mechanism of fermentation was. It was Louis Pasteur who demonstrated that fermentation requires living micro organisms like yeast cells. Buckner extracted a juice from yeasts, which was still capable of catalyzing the fermentation reactions. Such catalytic components were earlier called, by Willy Kuhne (1878) as enzyme. Later, Sumner succeeded in isolating an enzyme called urease in pure crystalline form, since then a large number of enzymes have been isolated and identified. Their chemical composition, structure, functions and kinetic mechanisms have been elucidated.

http://www.extension.iastate.edu/
NOMENCLATURE
In earlier days, nomenclature of enzymes was based on the substrate on which they acted ex. Sucrase on sucrose, lipase on lipids, protease on proteins, etc. As more and more number of enzymes was discovered in different labs all over the world, the trivial names gave rise to a lot of confusion. In order to avoid confusion and ambiguity in giving names, an international society of enzymologists was established. Based on substrate and the kind of reaction they brought about, the enzymes were given names.
CLASSIFICATION
In fact, naming and classification was done together. According to the international body of enzymologists, all enzymes are basically grouped into six classes of enzymes – such as oxido-reductases, transferases, hydrolases, isomerases, ligases and lyases; further sub groups have been identified.
OXIDO REDUCTASES
Enzymes responsible for oxidation either by the addition of oxygen or removal of hydrogen or electrons are called oxidases. On the contrary, the enzymes which add hydrogen or electrons are termed as reductases. Most of the enzymes which bring about oxidation or reduction perform the reaction simultaneously; ex. Cytochrome oxidase, nitrate reductase, a-ketoglutamate dehydrogenase.
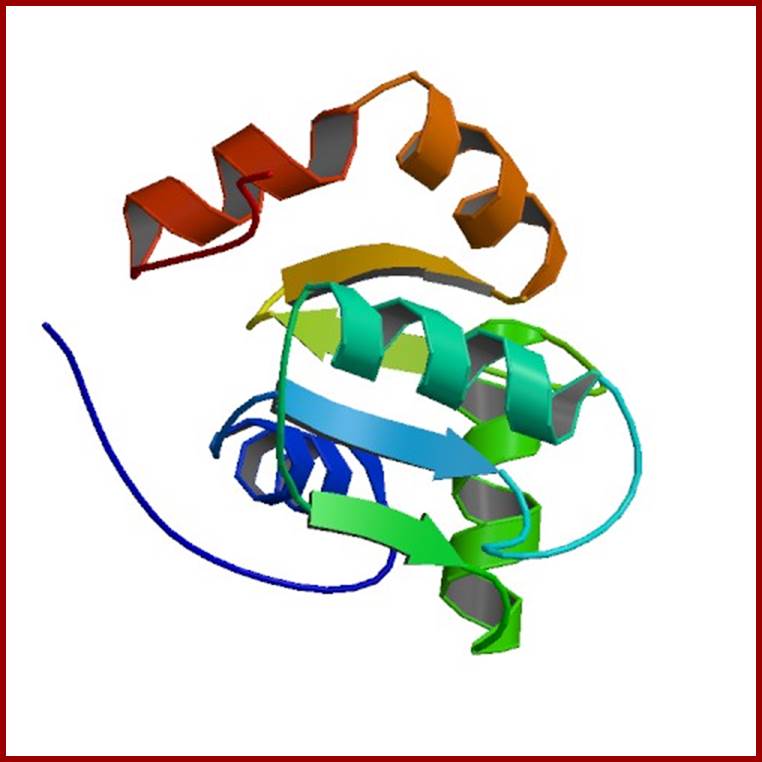
Glutaredoxin 5; http://www.rcsb.org/
TRANSFERASES
Certain groups like amino groups, acyl groups, phosphates, etc can be transferred from one compound to another compound by specific transferase enzymes, ex. Aminotransferases, transcarboxylases, transhydrogenases, transacetylases etc.
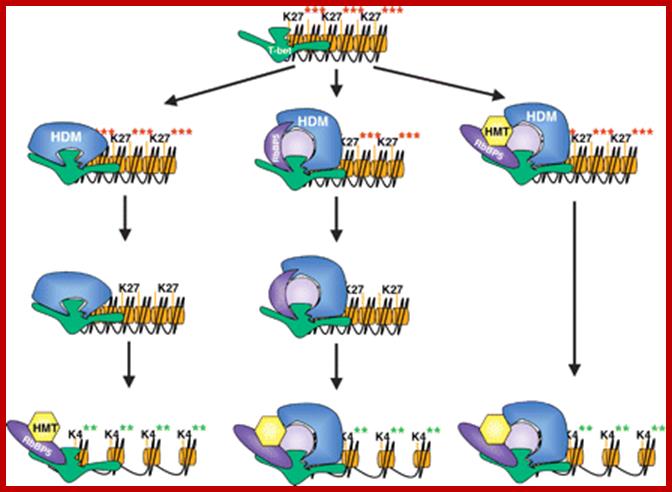
H3K27 Methyl transferase and demethylase; METTL7A; http://genesdev.cshlp.org/
Catechol O-methyl transferase domain, http://www.ebi.ac.uk/
HYDROLASES
The enzymes which by adding water molecules bring about breakdown of bonds are called hydrolases. Majority of lysosomal enzymes are hydrolases of one or the other kind, e.g. Lipases, proteases, DNase, RNase, Amylase, endonuclease etc.
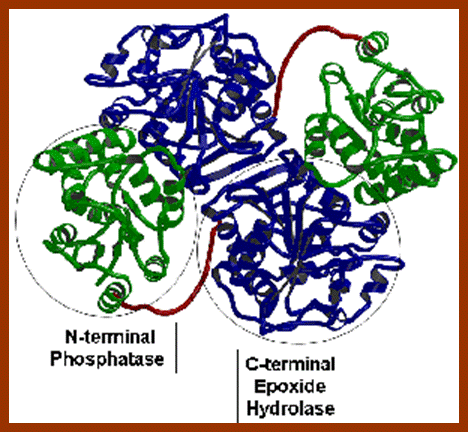
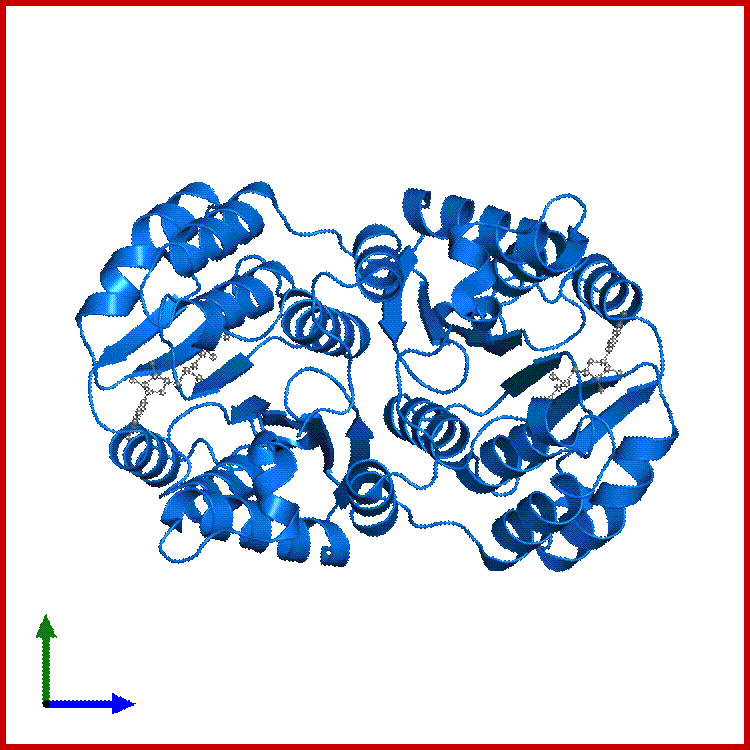
Epoxide Hydrolases; http://www.biopestlab.ucdavis.edu/
Pectin Lyase B; http://www.pnas.org/
LYASES
These enzymes add a group to compounds containing double bonds between carbon and carbon, carbon and nitrogen, carbon and oxygen, etc,
Adenylosuccinate Lyase; https://www.ebi.ac.uk
ISOMERASES
Enzymes which are capable of transforming one isomer to another are called isomerases. They a re highly specific in their substrates and reactions, ex. Glucose 6-p isomerase converts glucose 6-p to fructose 6p. Phosphoglyceraldehyde can be converted to another isomer called dihydroxyacetone phosphate by gluoco-phospho glyceraldehyde isomerase.
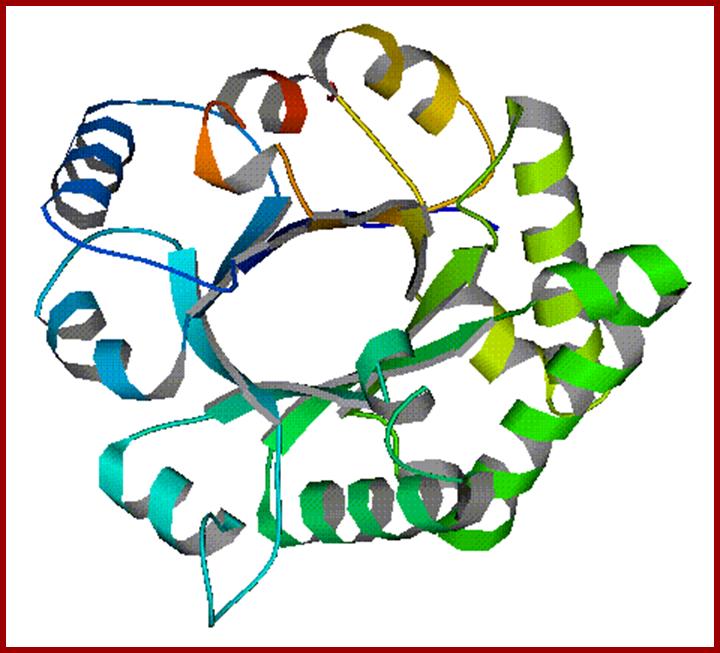
Triose phosphate Isomerase; http://www.mrc-lmb.cam.ac.uk/
LIGASES
Ligase enzymes bring about the bond formation between different molecules by removing a molecule of water. In fact, their activities is in the opposite direction of hydrolyses. They bring about the synthesis of bigger compounds by the addition of simpler compounds. Such enzymes are also called synthetases, e.g. RNA polymerases, DNA polymerase, peptidyl transferase etc.
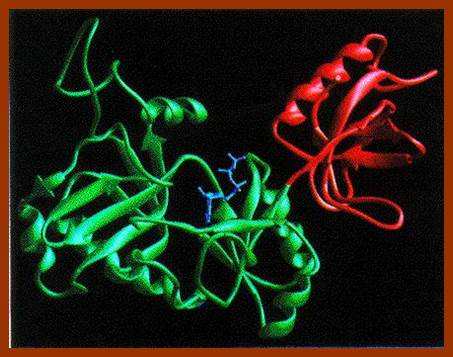
T7 DNA ligase; http://www.biochem.umd.edu/
CHEMICAL COMPOSITION AND STRUCTURE OF ENZYMES
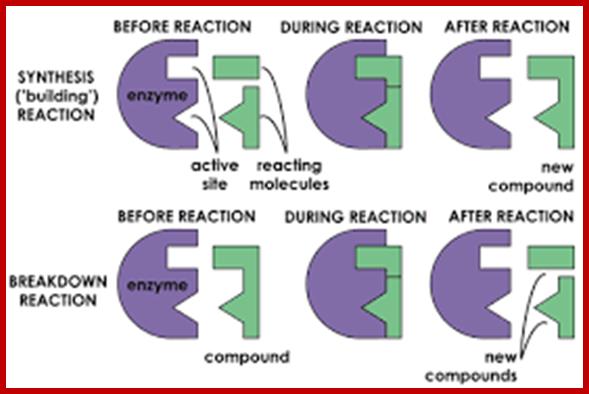
Almost all enzymes are made up of proteins as the major component. In addition some non proteinaceous compounds like vitamins or inorganic ions are also bound to be the protein part of the enzyme. The protein part of the enzyme is called ‘apoenzyme’ and the non protein parts are called as prosthetic groups, which may be metal ions like Mg2+, Ca2+, Mo2+, Mn2+, Fe2+, Cu, K, Ni, Se, Zn, etc. The apoenzyme may also contain some non proteinaceous organic compounds like NAD, FAD, NADP, FMN, coenzyme A, Biotin, TPP, Folate, pantothenic acid, etc. Such compounds are called co-enzymes. They easily dissociate from the main enzyme. The apoenzyme and prosthetic groups can be separated by dialysis; where the prosthetic groups diffuse out of the membranous bag, but the bulky apoenzyme protein part is retained within the dyalytic membranes. The enzyme containing both apoenzyme and prosthetic group together is referred to as Holoenzyme.
The main part of the enzyme is protein; it possesses a 3-D structural organization. The total surface area of the enzyme is very large, and it has specific sites at which the substrates bind to the enzyme. The apoenzyme may be made up of a single protein or it may consist two or more monomers. Still their organization and association is very important for their specific function.
However, some multiple enzyme systems, where two or more different enzymes are complexed together; and together they function. Such enzymes perform multi step reactions where intermediate products are retained on the enzymatic surface and only the final products are released from the surface of the multiple enzymes, ex. Pyruvate dehydrogenase, ketogluterate dehydrogenase, Fatty acid synthetase, etc.
The specificity of an enzyme and substrate and its function is determined by the enzymatic proteins’ 3-D organization. The enzyme proteins are folded in such a way, they possess specific regions in the form of clefts or grooves of particular shape and dimensions. The surface area of such sites is always complementary to their substrates, so that the substrate and enzyme form a complex similar to that of lock and key association. Within such grooves or clefts, certain amino acids with specific R groups act as binding sites to which the substrates bind transitorily and exert forces to bring about reactions.
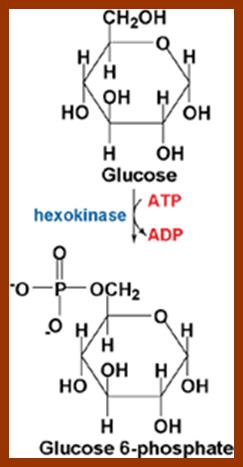
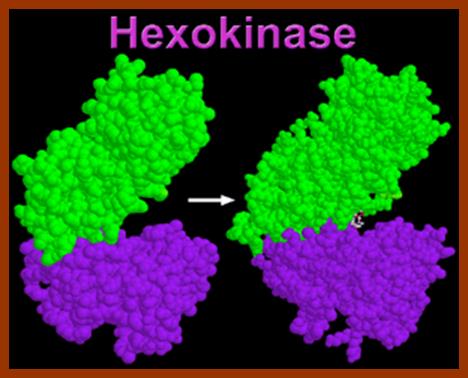
Hexokinase, the pacemaker of glycolysis; http://www.wiley.com/
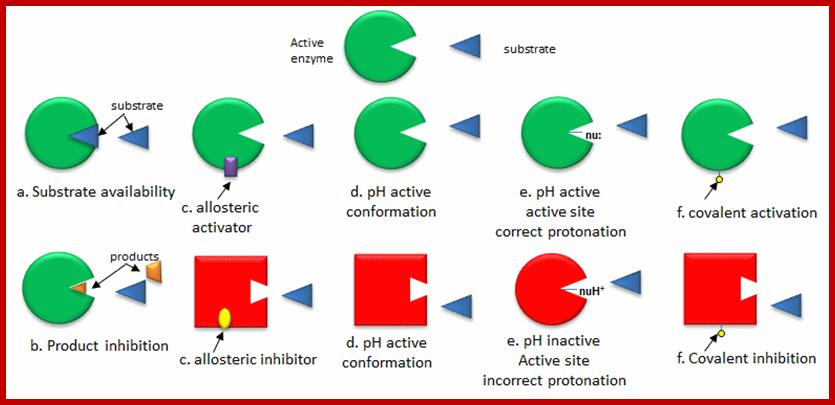
Some of the enzymes, besides possessing specific binding sites for substrates, contain other sites at which certain molecules bind and bring about conformational changes in the 3-D confirmation of the enzymatic protein. Such bindings may activate an enzyme or inhabit the activity of the enzymes. Such enzymes are called allosteric enzymes. The components that bring about the activation or inactivation of allosteric enzymes are called allosteric affectors or effectors. The affecters block the activity and effectors activate the activity.
Distribution of Enzymes
Multi-cellular organisms are made up of different organs containing specific tissues. Each of then perform a set of functions by producing specific enzymes. However, some of the enzymes synthesized within the cell are secreted to extra-cellular surfaces, such enzymes are called exoenzymes and those enzymes which are retained within the cell are called endoenzymes.
The enzymes present within a cell show a great range in their structures and functions. However, some of the enzymes are compartmentalized, so that each organ contains a group of enzymes which perform specific functions. For example, chloroplast contains enzymes responsible for photochemical and carbon pathways. Nucleus possesses enzymes responsible for DNA replication, transcription, processing, etc. Likewise, cytosol contains enzymes for glycolysis, and many metabolic pathway enzymes and many others. Thus one can see intracellular compartmentalization of enzymes is for specific functions.
CONSTITUTIVE AND INDUCED ENZYMES
A large number of enzymes present in cells are continuously made irrespective of external or internal conditions. Such enzymes are called house keeping or constitutive enzymes. But under certain conditions, specific enzymes are synthesized de novo. Such enzymes are called induced enzymes.
Properties of Enzymes:
1. Enzymes are macromolecules and possess a large surface area with specific binding or acting sites.
2. Enzymes act as biological catalysts
(a) They increase or accelerate the rate of reaction towards equilibrium
(b) Enzymes are required in small numbers to bring about maximum rate of reaction.
This is determined by the turnover reactions. The total number of products produced by a given enzyme in a given time is called turnover number. Different enzymes exhibit different turnover rates.
|
Substrate molecules transformed into products per enzyme/min/ |
|
|
Phosphoglucomutase |
1,240/min |
|
Glycosidase |
12,500/min |
|
Carbon unhydrase |
36,000/min |
|
Catalase |
56,00,000 |
|
Amylase |
11,00,000 |
|
Succinic dehydrogenase |
1,150 |
(c) Enzymes remain unaltered at the end of a reaction. Once the reaction is brought about, enzymes are ready for another sequence of reactions.
(d) Majority of the enzymes are capable of bringing about reversible reactions depending upon the concentration of substrates or products. If the concentration of substrates is more than the concentration of products, the enzyme favors forward reaction. On the contrary, if the concentration of substrates is less than the concentration of products, the enzymes favor reverse reactions. However, not all enzymes are capable of bringing about reversible reactions and they exhibit unidirectional reactions.
(e) The catalytic activity of enzymes is highly specific in terms of their substrates and the kind of reactions they bring about.
(I). General Specificity: Enzymes like RNAase degrade RNA molecules of every kind. DNAases digest all kinds of DNA molecules irrespective of their nucleotide sequences.
(ii) Absolute group specificity: Trypsin and pepsin are proteolytic enzymes, but they cleave peptide bonds at specific amino acids, ex. Trypsin is specific to the carboxyl side of arginine or lysine. Pepsin is specific to amino acids of tyrosine or phenylalanine residue in the protein.
(iii) Stereo chemical specificity: There are enzymes which recognize only specific isomers with either α or β or D or L forms. Such enzymes are called stereo specific enzymes, which are capable of transforming one isomer to another or vice versa.
The binding forces responsible for such an association may be due to metal ions, hydrophobic interactions or ionic interactions.
αβδ or B
3. Sensitive to Heat:
Enzymes being mainly made up of proteins, their 3-D organization depend upon S-S bonds. Under normal temperatures such bonds are intact and perform normal functions but at very high temperatures, the S-S bonds break open and proteins get denatured and their function is impaired. Further more, the rate of reaction depends upon the temperature. As the rate of reaction depends upon the frequency of collision between the substrate and enzymes, the rate of movement of these reactants is controlled by the kinetic energy available in the system.
4. Sensitive to pH:
As enzymes are mainly made up of proteins, the electronic charge of the R-Groups found in amino acids depends upon the intracellular pH. Every enzyme has an optimal pH for its activity. Quite a number of enzymes are active at the range of pH 5.8 to 6.8. But certain enzymes like Trypsin and chymotrypsin are active at acidic pH 3-4. On the contrary, Alkaline phosphotases are active at pH 10-12. As pH of the cytoplasm determines the activity of functional groups found in enzymes, their activity is dependent on specific pH.
5. Sensitive to Inhibitors:
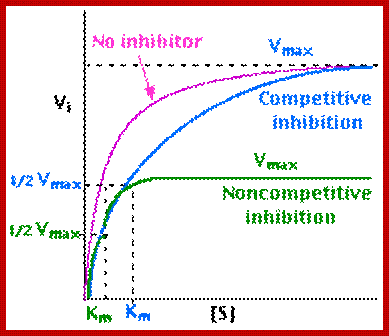
Normally substrates bind to enzymatic surface at specific sites, before catalytic action. But certain molecules other than substrates sometimes bind to active site or at some other site and bring about the inhibition of enzymatic activity. Such substances are called inhibitors, which may be competitive, non-competitive or un-competitive inhibitors.
1. Competitive Inhibitors:
Competitive inhibitors are those molecules, whose structural configuration is almost similar to that of actual substrates. As enzyme active sites recognize certain specific groups found in the substrate, any inhibitor molecule which possesses such groups identical to a substrate easily binds to active site. But the enzyme fails to bring about any catalytic reactions because of the internal structure of the inhibitors. Thus competitive inhibitors, by binding to active sites prevent the binding of substrates to the enzymatic surface. Competitive inhibition could be reversed by the addition of excess amount of substrates. For example, succinate, a substrate binds to its enzyme called succinate dehydrognase. If malonate, which has carboxyl groups similar to that succinate, is added, it easily recognizes the active sites in the enzyme and prevents the binding of real substrates. Thus enzyme activity is inhibited.

http://chemistry.elmhurst.edu/http://chemistry.elmhurst.edu/
2. Non Competitive Inhibitors:
Certain organic or inorganic molecules inhibit enzymatic activity by distorting the 3-D surface of the protein or by blocking the active site non-competitively. The former kinds of inhibitors bind to a site of an enzyme other than active site and induce conformational change in the structure of proteins, thus making the enzyme inactive. On the other hand, compounds like cyanide, rotenone, antimycin, etc. bring to the active site of respiratory enzyme non-competitively and inhibit enzymatic activity. There are another class of compounds like Urea, Mercaptide compounds which break the S-S bonds and unfold the proteins and make it inactive.
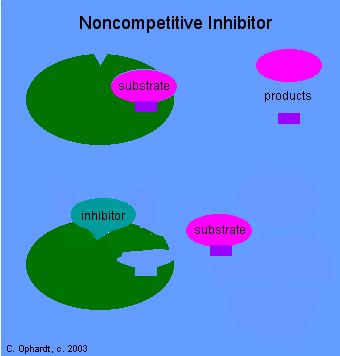
Noncompetitive Inhibitor-http://chemistry.elmhurst.edu/
EnzymeKinetics.html; http://users.rcn.com/jkimball
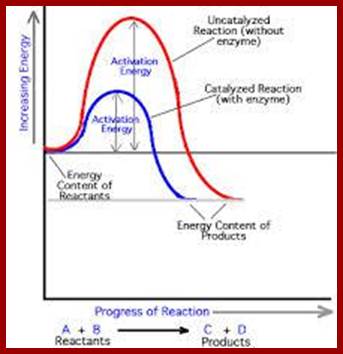
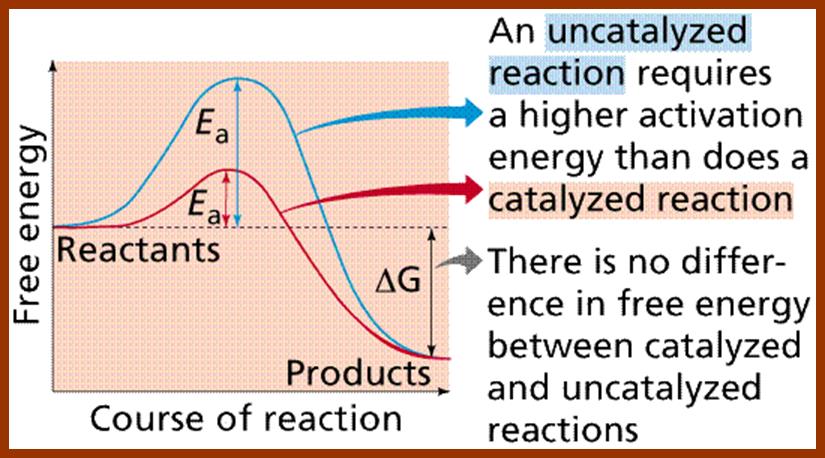
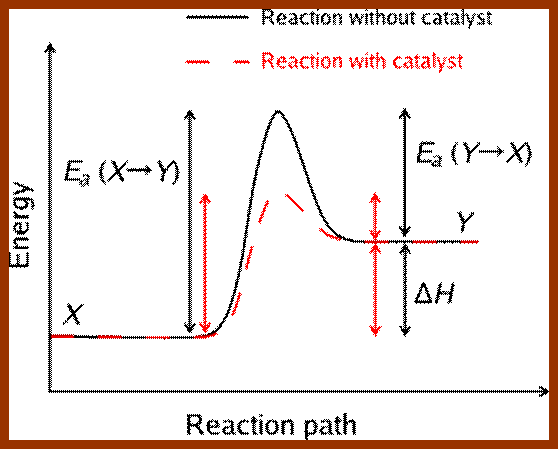
6. Lowers the Activation Energy:
In living systems, molecules are under constant motion and exist at different energy states. Some may exist at higher energy state and some exist at may lower energy start. Without the mediation of enzymes, substrates by themselves can react with each other by collision, provided the energy required is sufficiently high, at which point, molecules are in a transitory state. The amount of energy required for a substrate to be at higher transitional state is referred to as activation energy. For example, sucrose breaks down spontaneously, if the activation energy available is of 28000 K.cals/mole. But in the presence of enzyme invertase; it performs the same reaction with just 11000 K.cals/mole. Thus enzyme medicated catalysis requires less energy, because the binding of substrates to enzymes, which have a large surface, renders substrate molecules to be in a transitory state, because binding brings about stretching of the substrate bonds. This greatly felicitates the reaction with minimum input of energy. Thus enzymes economize the utilization of cellular energy and also make them efficient.
https://suckasaccharide.wordpress.com/
http://www2.estrellamountain.edu/
http://www.yale.edu/ynhti/
Enzyme catalysis requires less energy, because the binding of substrates to enzymes, which have a large surface, renders substrate molecules to be in a transitory state, because binding brings about stretching of the substrate bonds. This greatly felicitates the reaction with minimum input of energy. Thus enzymes economize the utilization of cellular energy and also make them efficient.
Enzymes conserve energy during their course of reactions. http://www1.lsbu.ac.uk/
ISOZYMES
An enzyme can exist in two or more different structural forms still perform the same functions. Such enzymes are called isoenzymes. Such behavior is due to changes in the amino acid composition of enzymatic proteins, ex. Lactic dehydrogenase, it is a tetramer and it exists in five different forms; which are found in different organs. Such multiple forms of enzymes may be expressed at different stages of development. Electrophoretic methods have greatly helped in identifying such isoenzymes.
PRECURSOR ENZYMES
In recent years, studies on biosynthesis of macromolecules like polypeptides revealed that most of the proteins that are synthesized on mRNA template are larger than the functional proteins. Such large inactive proteins are called precursor proteins or they may also exist as pre pro precursor proteins. Such proteins require enzymatic cleavage of certain part of the polypeptide chain for the activation of precursor proteins. Living organisms produce some inactive enzymes called zymogen granules, ex. Chymotrypsinogen, Trypsinogen, pepsinogen, etc. Chymotrypsinogen is activated into active Chymotrypsinogen by enzymatic cleavage of a peptide bond. Similarly trypsinogens and pepsinogen are converted to active Trypsin and pepsins by enzymatic activation. Even insulin proteins are first synthesized as inactive precursor proteins, they are then made active by elimination of a particular segment of the protein chain.
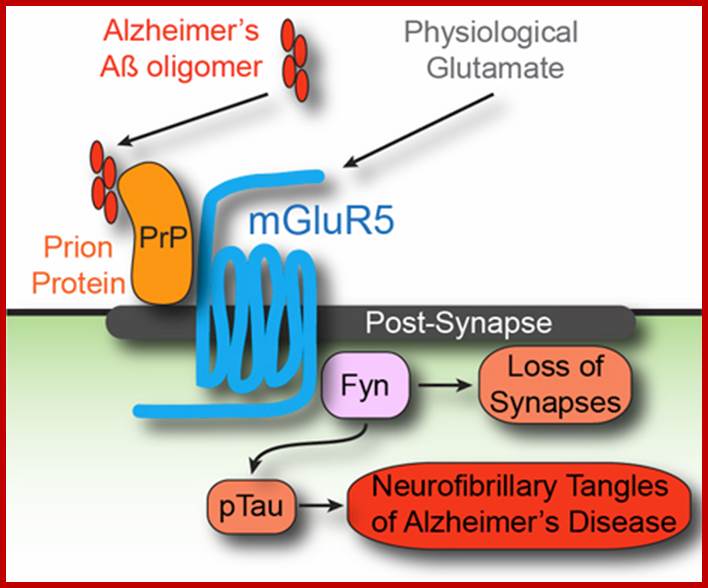
The complete Alzheimer’s pathway, with the uncovered mGluR5 G-Protein Coupled Receptor. Courtesy of Professor Stephan Strittmatter; http://www.yalescientific.org/
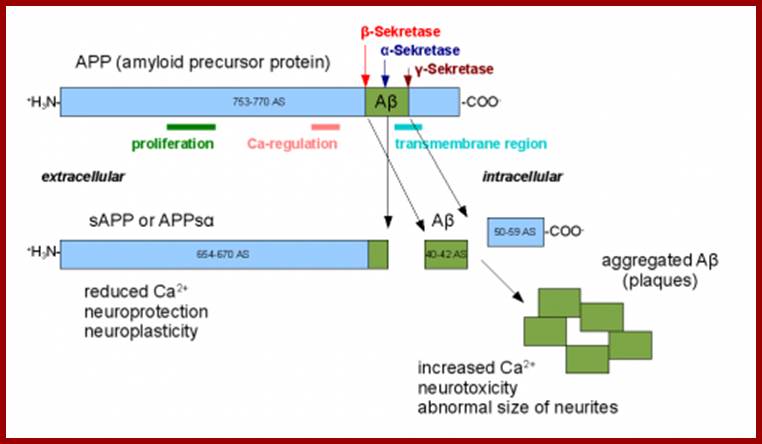
Amyloid beta precursor protein, when cut by the incorrect secretase enzyme, can form amyloid beta plaques, leading to Alzheimer’s disease. Courtesy of Professor Joseph Wolenski. ;http://www.yalescientific.org/
MECHANISM OF ENZYMS ACTION
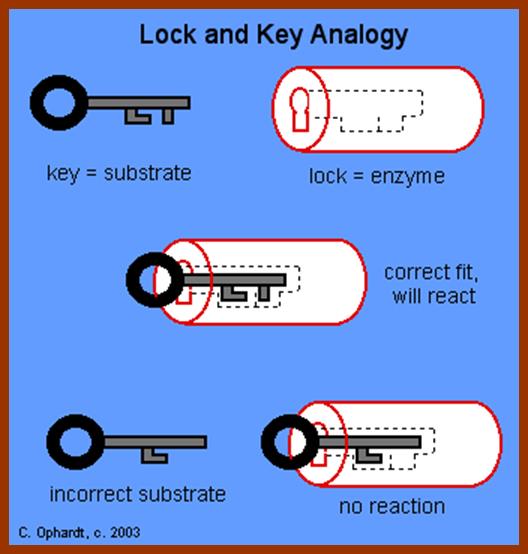
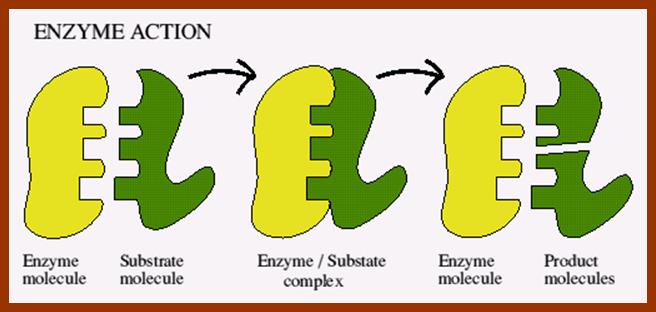
In recent years, various techniques like radioactive isotope labeling, spectrophotometric, immunoprecipitation techniques, etc. have been employed in understanding the kinetic properties and the mechanism of enzyme action. Way back, Emil fisher proposed lock key model to explain the enzyme substrate reactions. However, this model has been slightly restructured to explain certain properties of enzymes.
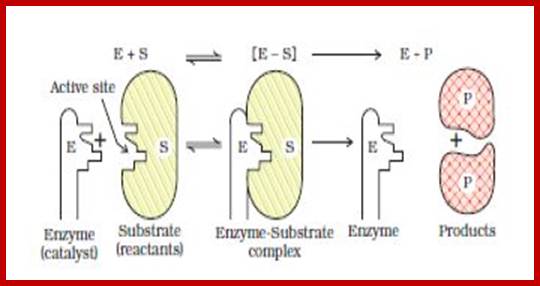
LOCK AND KEY THEORY
To going with the enzyme and substrate molecules collide with each other. If the collision brings their complementary surfaces together, the electronic forces operating upon the enzymes and substrate molecules, facilitate the binding of substrate to the active site located on the enzymatic surface. The active site is always located in a cleft or a groove within the enzyme.
http://www.slideshare.net/
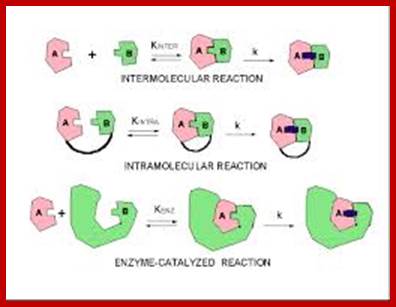
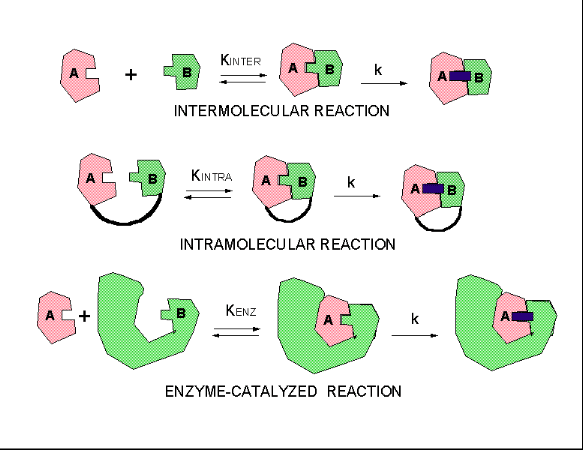
Three kinds of reactions, intermolecular, intramolecular, and enzyme-catalysed, above diagram; http://employees.csbsju.edu/ https://biochemaliensunite.wordpress.com
The binding, is mostly due to non covalent forces like hydrogen bonding and it is a transitory phenomenon. The R groups in amino acid residues found in the active site exert many forces like Vander wall forces, hydrogen bonding, ionic interactions, or hydrophobic interactions.
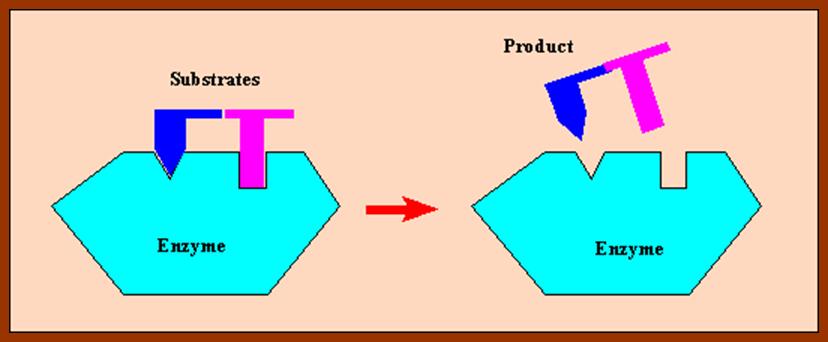
 T
T
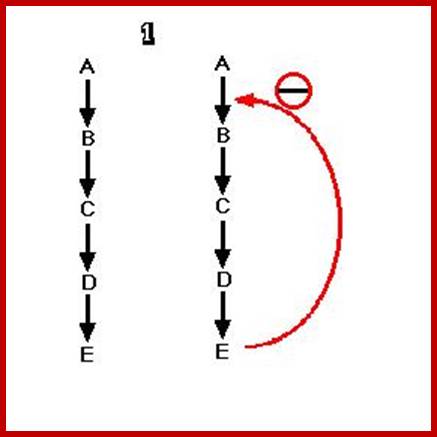
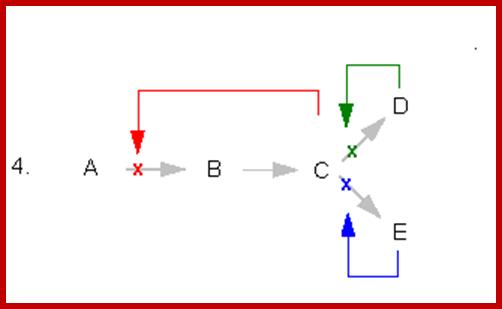
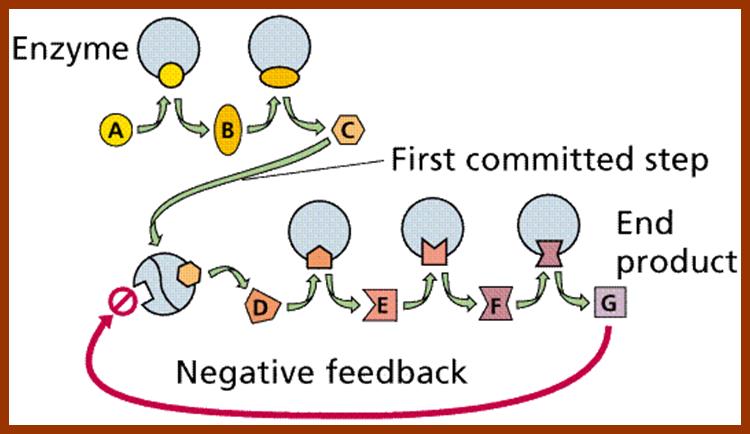
This figure illustrates simple feedback inhibition. If the end product of the metabolic pathway (E) accummulates it inhibits the activity of the first enzyme in the pathway. Examples can be found in amino acid biosyntheses. The enzyme affected will be an allosteric enzyme. http://biocadmin.otago.ac.nz/
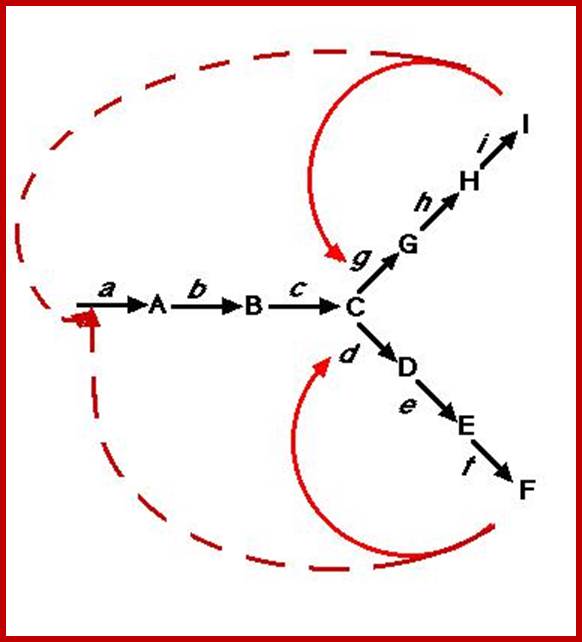
http://biocadmin.otago.ac.nz/
Control
in a branched pathway. An extension, in terms of complexity, of the above;
1) Feedback inhibition can occur after the branch point as in Figure 1 (solid
arrows).
2) Control prior to the branch point (broken arrows) can be effected in a
number other ways, in this example it can be by :
i) Concerted Feedback Inhibition. In this case both end products are required
to b bound to effect inhibition (two different allosteric sites).
Other strategies that are found:
ii) Cooperative End product Inhibition, where either end product is weakly
inhibitory but together they are more than additive in their inhibitory effect,
and
iii) Cumulative End product Inhibition, where each end product inhibits the
enzyme by a given amount irrespective of the presence or absence of other end
products (effects not more than additive).
Feedback Inhibition; http://employees.csbsju.edu/hjakubowski
https://s10.lite.msu.edu
https://biochemistry3rst.wordpress.com
Enzymes-Advanced Level; http://fwdssp.com/Science
https://rxize.wordpress.com
http://chemistry.elmhurst.edu/
http://www2.estrellamountain.edu/
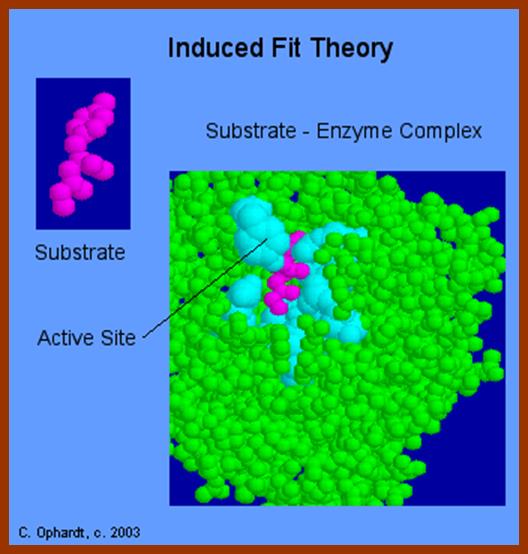
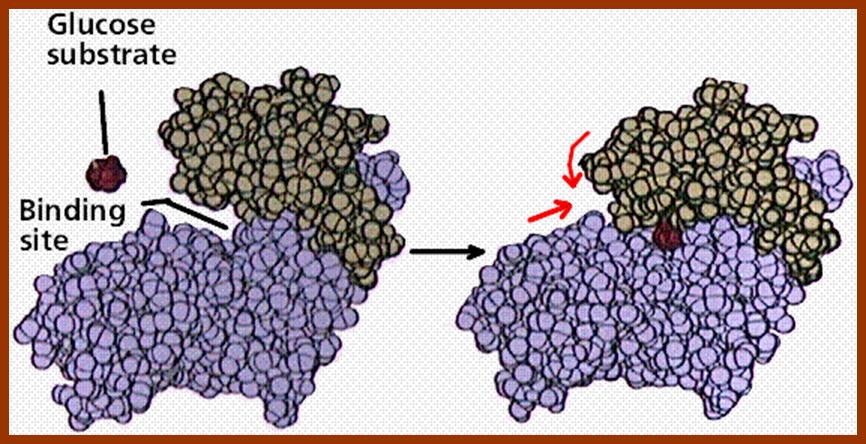
The initial binding of a substrate induces certain changes in 3-D conformation of the enzyme, which felicitates the substrate to bind to the enzyme properly, what is called ‘face to face’ binding. Such changes in enzyme topography by substrate were first proposed by Koshland Jr. This mechanism is popularly called as ‘Induced fit mechanism’. It is now believed that most of the substrate enzyme binding reactions are found to be induced fit types.
http://fantasticsams.wikispaces.com/
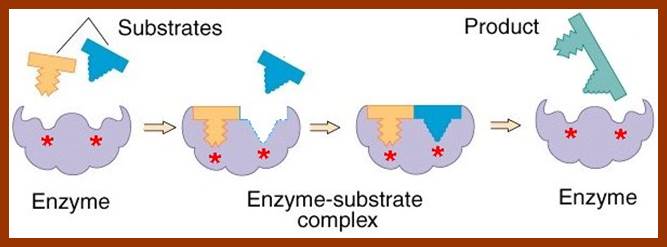
The proper binding of the substrate to the enzyme renders the substrate to be in a transitional state. The electronic forces operating in the region of active site bring about the reaction either in making of a bond or breaking off a bond or involving in the transfer of a group. The reorientation of bonds within a substrate molecule, brings about and change in the configuration, hence it becomes a product. As products are in a stable form, they are repelled and released from the surface of the enzyme and make the enzyme free for another cycle of reactions. In the said mechanism, a single substrate binds to a single enzyme and the products produced may be one or two.
![enzyme; active site [Credit: Encyclopædia Britannica, Inc.]](Plant_Cell_Biochemistry_And_Metabolism1-Proteins_And_Enzymes_files/image163.jpg)
http://www.britannica.com/, science/enzymes
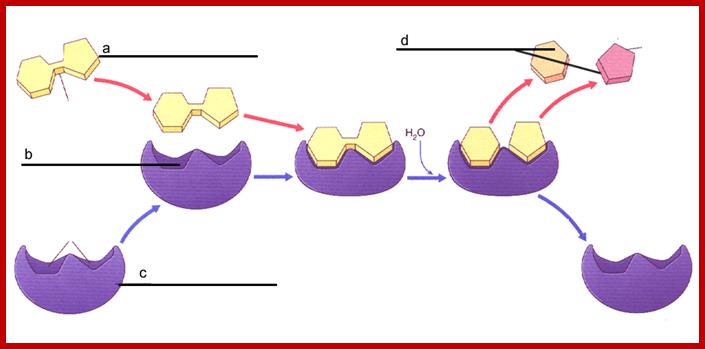
But there are many enzymatic reactions, where two substrates are involved to produce one or two products. In such cases the mechanism is slightly different. They are single displacement mechanism and double displacement or ping pong mechanism.
SINGLE DISPLACEMENT REACTIONS
These reactions involve the binding of two substrates simultaneously to two specific binding sites on the enzyme. Binding leads to enzymatic reactions and then the product are released from the enzyme surface.
DOUBLE DISPLACEMENT OR PING PONG REACTIONS
Ping-Pong reactions are called double displacement reactions. Tis is also called as Bisubstrate reactions. In these reactions one of the two substrates binds to a specific site, first. The enzymatic reaction that ensues, results in the transfer of group from the substrate onto the enzyme surface and the substrate after donating the groups is released as the product. Then the second substrate binds to the second site at which a group found on the enzyme is transferred to the substrate and the product is released.
E + A EA E'P E' + P
Then:
E' + B E'B EQ E + Q
A "ping pong" or "double displacement" bisubstrate reaction; http://www.mikeblaber.org/
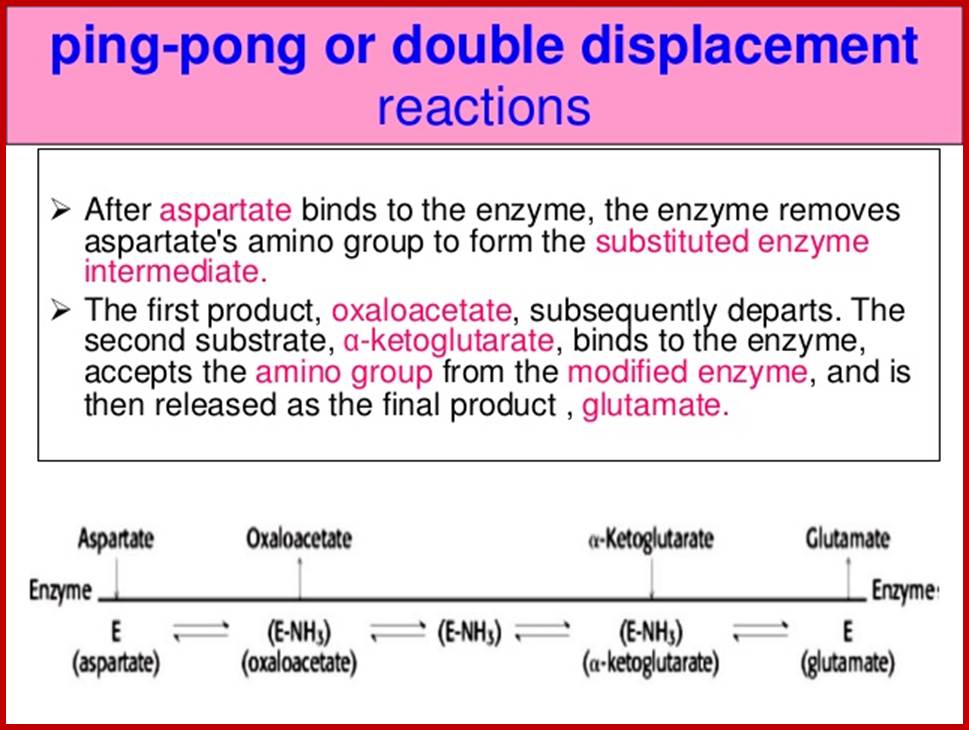
Double displacement reactions; http://www.slideshare.net/
Random single displacement reactions; http://www.mikeblaber.org/
- All combinations of enzyme, substrate and product are possible:
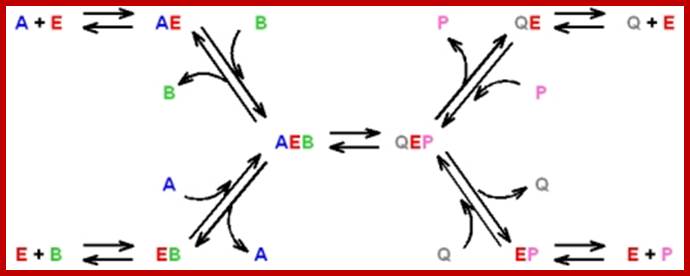
- The rate-limiting step is the catalytic step: AEB QEP
http://www.mikeblaber.org/
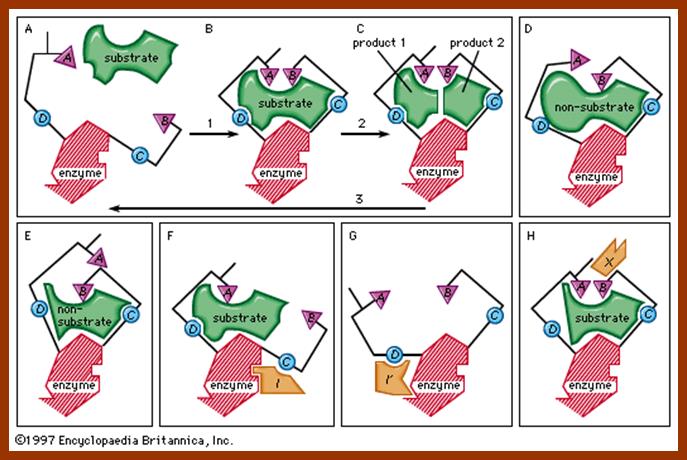
EVIDENCES FOR ENZYME SUBSTRATE COMPLEX FORMATION
Various methods have been employed to find out whether enzyme and substrate molecules bind to each other to bring about enzymatic reactions. Only spectrophotometric, immunoprecipitation and chemical modifications have been mentioned here.
Enzyme and its active site for the binding of substrate.http://www.mun.ca/

Enzyme-substrate complex; http://www.biology.arizona.edu/biochemistry
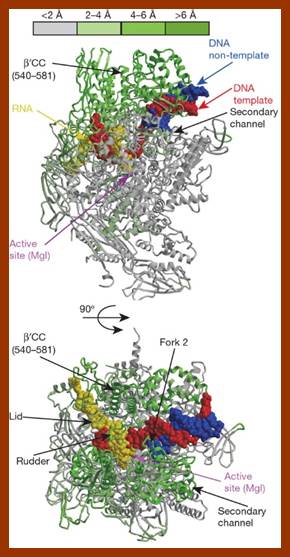
{kind=link}
{kind=link}
{kind=link}
Enzyme and substrate DNA complexed in the reaction
SPECTROPHOTOMETRIC METHOD
Different organic substances in their free state absorb light at different wavelengths, and show distinct absorption spectrum. So the enzyme and substrate individually show distinct absorption spectrum, which can be easily determined by spectrophotometric studies. If an enzyme and a specific substrate are bound to each other, the absorption band shifts. This does not happen, if the enzyme and the substrate are free.
IMMUNOPRECIPITATION METHOD
Enzymes can be immunoprecipitated by using specific antibodies. If labeled substrate is added to an enzyme solution and immediately immunoprecipitated, the substrate that just bound is also precipitated along with the enzyme. Thus the enzyme substrate complex formation can be demonstrated.
CHEMICAL MODIFICATION
As enzymes contain specific binding sites, they can be modified chemically or they can be made inaccessible for the substrate and thus reaction can be inhabited. This is another evidence to prove that substrates bind to enzymes at specific sites.
ENZYME KINETICS
Different events like enzyme to substrate contact, binding, reaction and the release of products are time consuming processes. The time required for a given enzyme to bind and convert the substrate to its products can be determined by various methods and by different means. However, the rate with which enzymatic reactions taken place is referred to as enzyme kinetics. The relationship between the enzyme and substrate and other factors operating in the system can be determined accurately by kinetic data. The rate of reaction is always measured in terms of conversion of a number of substrate molecules into products by a single enzyme molecule, in a given time.
Enzyme reactions follow the pattern of order of reactions such as; Zero order, First order, Second order, Zero order and Mixed-order reactions. http://www.chem.purdue.edu/
Zero order Reactions:
Zero-order reactions (order = 0) have a constant rate. This rate is independent of the concentration of the reactants. The rate law is: rate = k, with k having the units of M/sec.
The integrated rate law is [A] = -kt + [Ao]
First Order Reactions;
A first order reaction (order = 1) has a rate proportional to the concentration of one of the reactants. A common example of a first-order reaction is the phenomenon of radioactive decay. The rate law is: rate = k[A] (or B instead of A), with k having the units of sec-1
The integrated rate law is ln [A] = -kt + ln [Ao]
Second-Order Reactions;
A second-order reaction (order = 2) has a rate proportional to the concentration of the square of a single reactant or the product of the concentration of two reactants:
rate = k[A]2 (or substitute B for A or k multiplied by the concentration of A times the concentration of B), with the units of the rate constant M-1sec-1
The integrated rate law is 1/[A] = kt + 1/[Ao]
Mixed-Order or Higher-Order Reactions
Mixed-order reactions have a fractional order for their rate: e.g., rate = k[A]1/3
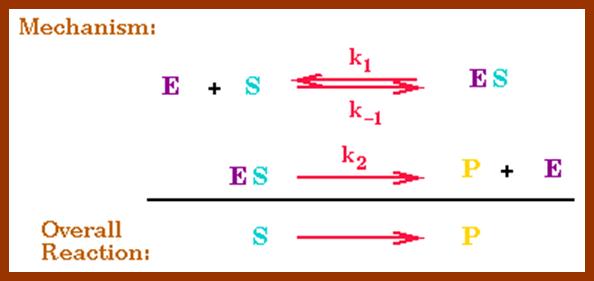
Leonor MICHAELIS AND MAUD LEONARA MENTEN EQUATIONs:
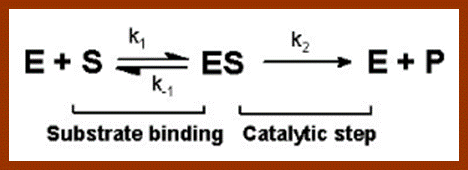
Using enzyme substrate complex formation as the criteria for enzymatic reaction, Michaelis and Menton developed an equation to explain the kinetic properties of various enzyme reactions. The quotation is very popularly called Michaelis Menton equation; it is possible to determine various quantitative aspects of enzymatic reactions like velocity, km value, effect of factors, temperature, inhibitors, etc.
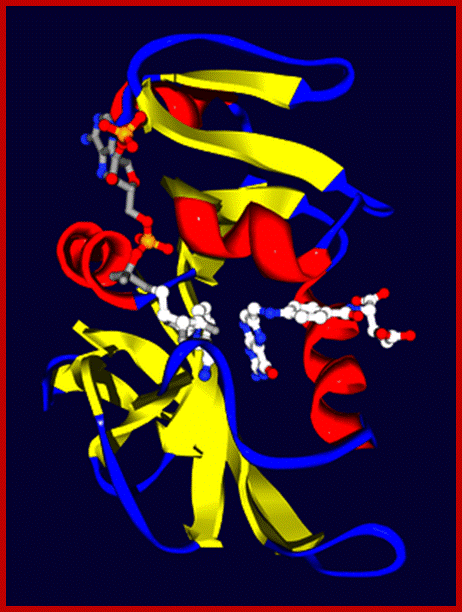
Di hydrofolate reductase from E.coli with ito substrates, dihydrofolate right and NADP left bound to their active sites; protein is shown in ribbon with alpha helices in red , beta sheets in yellow and loops in blue; https://en.wikipedia.org/wiki/Enzyme_kinetics
![]()
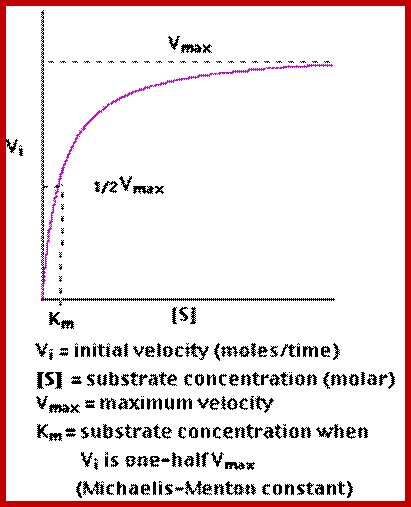
http://users.rcn.com/jkimball.ma.ultranet
The Michaelis–Menten equation[9] describes how the (initial) reaction rate v0 depends on the position of the substrate-binding equilibrium and the rate constant k2.

(Michaelis–Menten equation) with constants
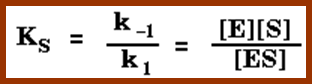


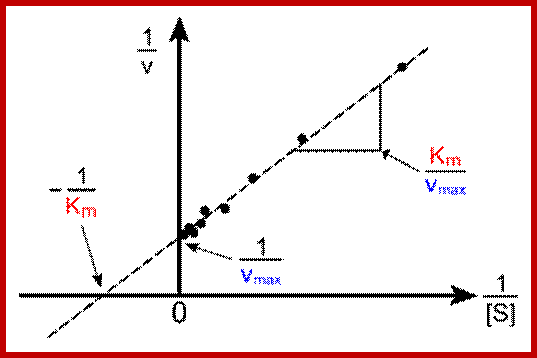

Lineweaver-burk’s reciprocal plot of kinetic data showing the significance of axis intercepts and gradient.
Plotting the reciprocals of the same data points yields a "double-reciprocal" or Line weaver-Burk plot. This provides a more precise way to determine Vmax and Km.
- Vmax is determined by the point where the line crosses the 1/Vi = 0 axis (so the [S] is infinite).
- Note that the magnitude represented by the data points in this plot decrease from lower left to upper right.
- Km equals Vmax times the slope of line. This is easily determined from the intercept on the X axis.
![]()
(Michaelis–Menten equation)




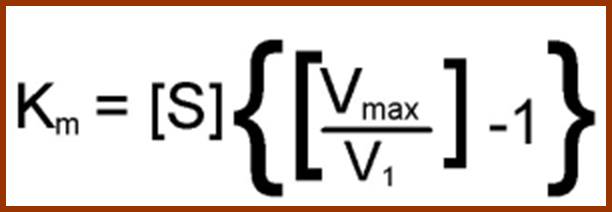
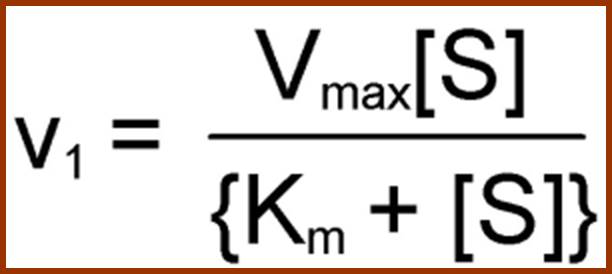
Vmax = K cat [ES] tot
![]()


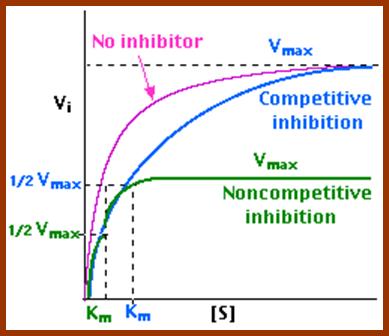
http://users.rcn.com/jkimball.ma.ultranet
In the presence of a competitive inhibitor, it takes a higher substrate concentration to achieve the same velocities that were reached in its absence. So while Vmax can still be reached if sufficient substrate is available, one-half Vmax requires a higher [S] than before and thus Km is larger.
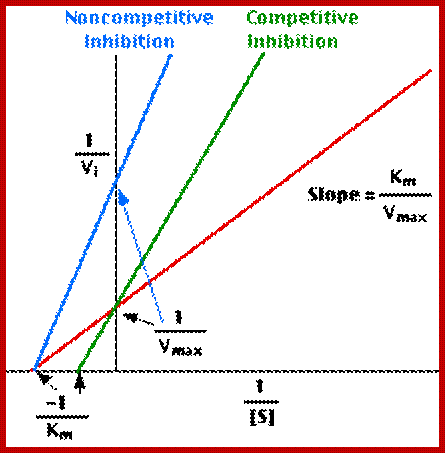
With noncompetitive inhibition, enzyme molecules that have been bound by the inhibitor are taken out of the game so
- enzyme rate (velocity) is reduced for all values of [S], including
- Vmax and one-half Vmax but
- Km remains unchanged because the active site of those enzyme molecules that have not been inhibited is unchanged.
This Lineweaver-Burk plot displays these results.
Assume that only one substrate molecule, binds to an enzyme to form an enzyme substrate complex, which results in the formation of products. The direction of the reaction is proportional to the concentration of input or the output. It is a reversible reaction.
In this equation, enzyme combines with a substrate to form a complex the complex produces a product. The rate constants for the forward reaction are K1, K2 & K3 where (E) (S) to (ES) and (ES) to (E) +(S) are reversible but from (ES) to (E) + (P) is not reversible.
E + S ==èES==èE + P
The velocity of enzyme reaction can be studied in individual reactions. V=K3 (ES) where the rate of enzymatic reaction is equal to the products of K3 and ES – (2)
The rate of formation of enzyme substrate complex is.
ES=K1 (E) (S) – (3)
The rate of breakdown of enzyme substrate complex to products is
ES = (K2 + K3) (ES) – (4)
Therefore K1 (E) (S) = (K2+K3) (ES) - (5)
The above equation can be further simplified or modified by using another constant called Michaelis Menton constant called km. In enzymatic reaction, km refers to the concentration of the substrate at which the enzyme reaction is half the V max. V max refers to the maximal rate of reaction at which all the enzyme molecules are saturated with substrates. The modified equation can be written as
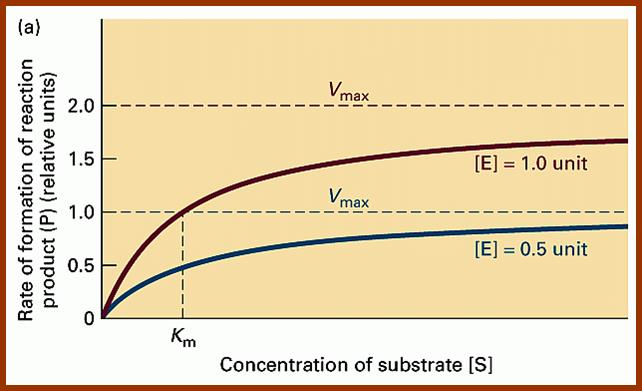
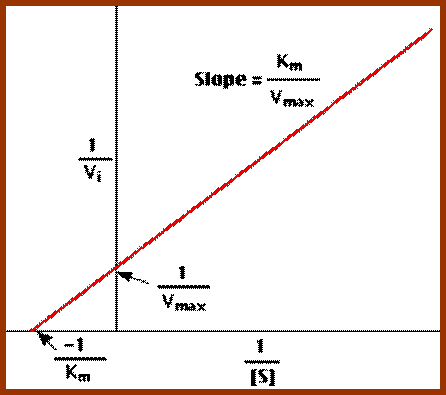
http://users.rcn.com/jkimball.ma.ultranet
Reciprocal plots:
The data obtained from Michaelis Menton equation can be plotted reciprocally as reciprocal of the velocity as the function of reciprocal of substrate concentration. The reciprocal plot can be used to determine the values for V max, km turnover numbers, etc. by just varying the concentration of substrates.
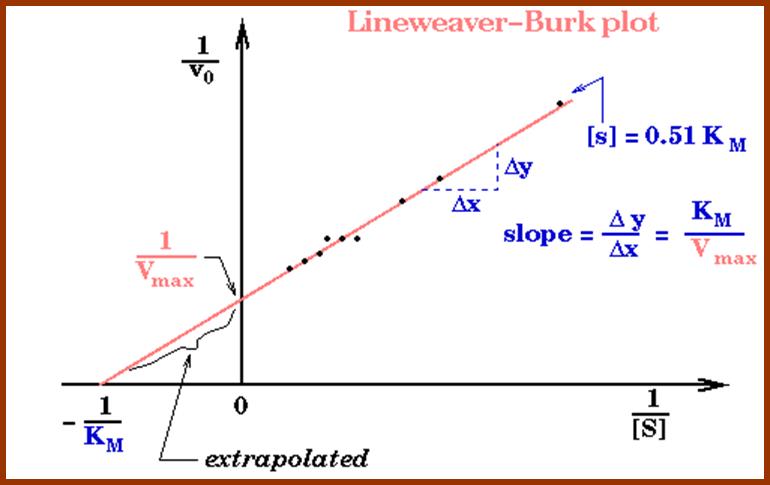
http://users.rcn.com/jkimball.ma.ultranet
Km of enzymes in mM concentration:
Catalase 25 mM
Carbonic anhydrase 26.0 mM
Hexokinase 0.4 mM
RUBP carboxylase 15 mM
RUBP carboxylase 1-2 mM
Turnover numbers, Kcat of some enzymes
Enzyme Substrate K cat (s-1
Catalase H2O2 40,000,000
Carbonic anhydrase HCO3 400,000
Acetyl choline esterase Ac-choline 14,000
B-Lactamase Penicillin 2000
Fumerase Fumarate 800
RecA ATP 0.4
Furthermore, determination of V max gives the data for calculating the turnover number for an enzyme. The number of substrate molecules converted into products per unit time when the enzyme is fully saturated with the substrate is called turnover number. Using this method, the turnover number of different enzymes has been determined. The reciprocal plot can also be used to find out the nature of inhibitors, whether they are competitive types or non-competitive types. See the figures.
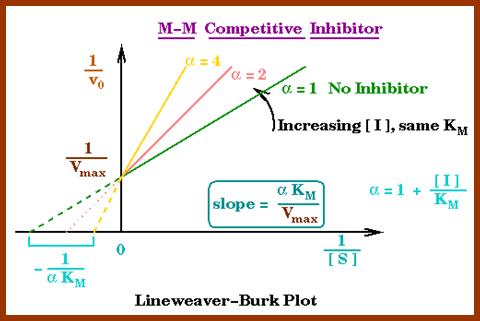
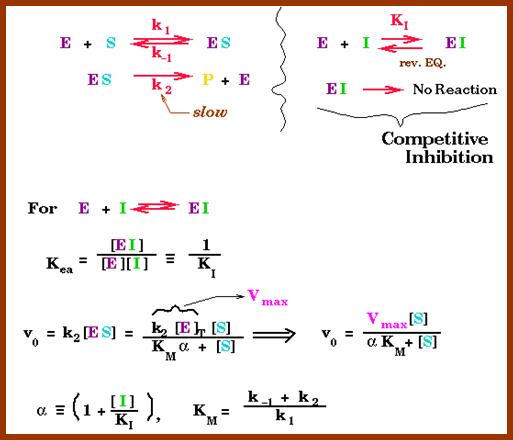
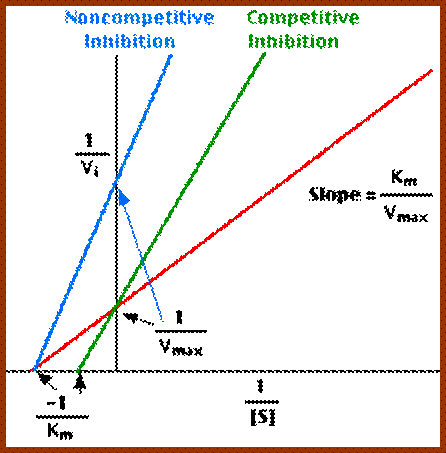
http://users.rcn.com/jkimball.ma.ultranet/
ALLOSTERISM AND ENZYME REGULATION
Enzyme with two or more sub-units is called multimeric or polymeric enzymes. Besides an active site, such enzymes contain another site at which molecules other than their substrates bind and bring about the modulation on the activity of the enzyme. Such sites are called allosteric sites and the enzymes are called allosteric enzymes. The phenomenon is referred as allosterism.
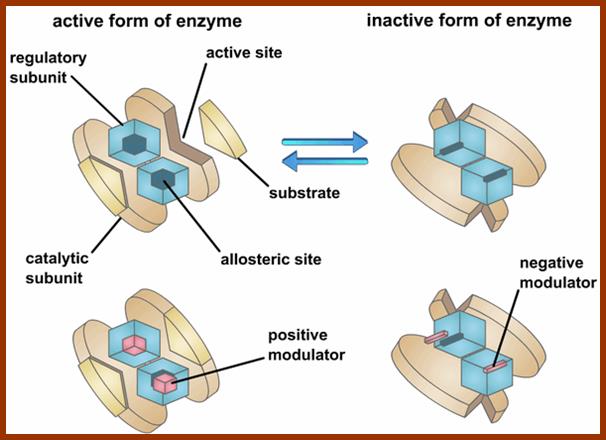
https://jkrishnan.wordpress.com
Certain molecules by binding to such allosteric sites induce a conformation change in the 3-D structure of the enzyme and make the inactive enzyme into an active enzyme. Such sites are called allosteric efforts or activators. Contrary to this effect some molecules may bring about the inhibition of enzyme activity, and then the said molecules are named as affecters or inhibitors.
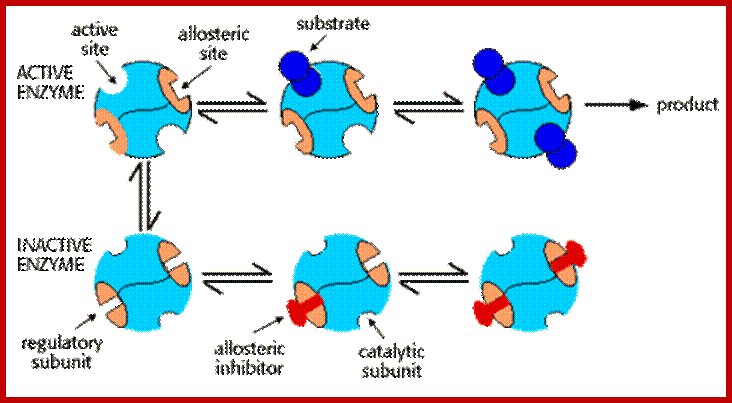
http://www.biology.arizona.edu/
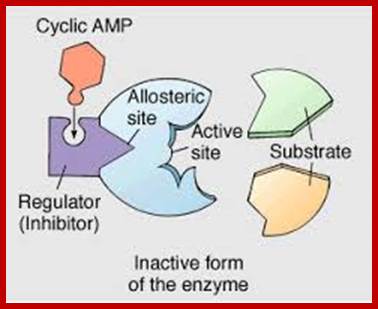
https://dopeahmeanbio.wordpress.com
In addition to the above said features, multimeric enzymes contain one or more binding sites for different substrates. The binding of one substrate to one of submits in the enzyme induces or felicitates the binding of the other substrates. This behavior is called co-operative effect. Thus one can observe how the enzymatic activity is modulated by various components like substrates, activators, inhibitors etc.
Cascade effect of signal binding to G-protein linked receptor; http://www.mun.ca/
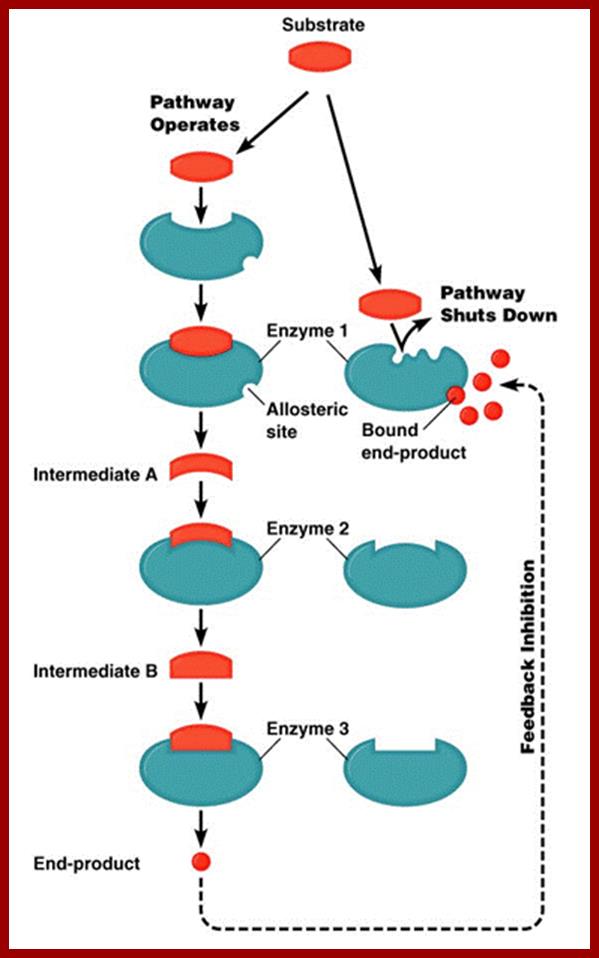
http://classes.midlandstech.edu/
In biological systems, there are many biochemical reactions where a particular substrate is converted to a product not in a single step but in multiple but sequential steps of reactions. And each of these steps is controlled by a specific enzyme. Many such multi step enzymes are induced enzymes. However, these reactions show very interesting features. The enzymes are induced by a specific substrate.
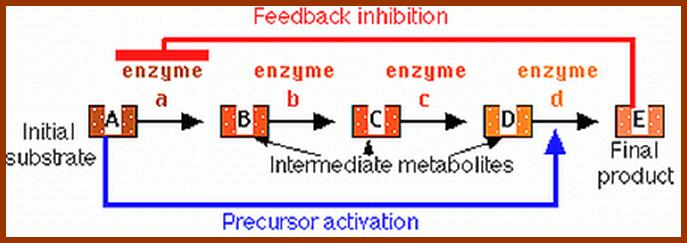
http://users.rcn.com/jkimball.ma.ultranet/
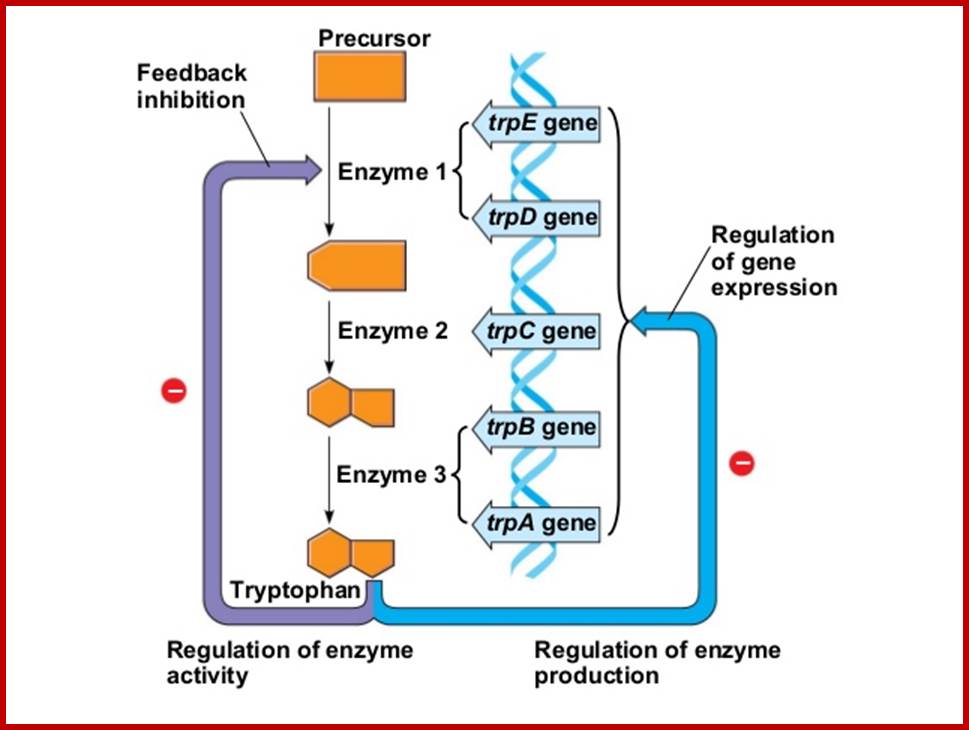
http://www.slideshare.net/
The product required synthesized when it is needed and the synthesis is stopped when it is not needed. In the latter mentioned control mechanism, the ultimate product, if it is excess, binds to the enzyme that controls the first step and makes it inactive, thus the production of final product is inhibited. Such a phenomenon is called feedback inhibition. Similar enzyme regulatory mechanism can be applied to a branched pathway.
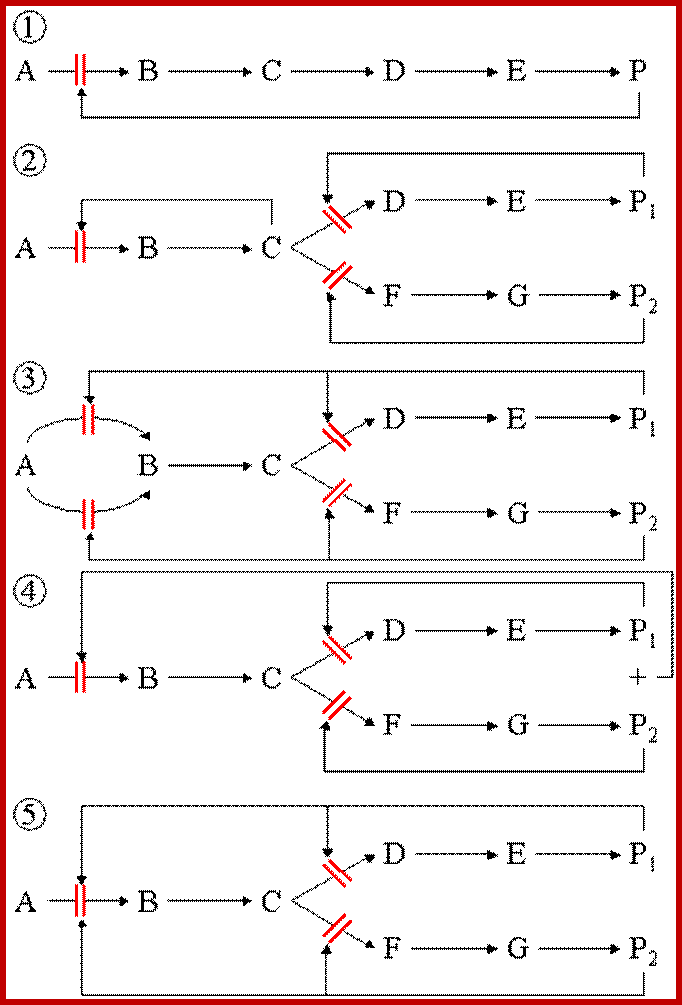
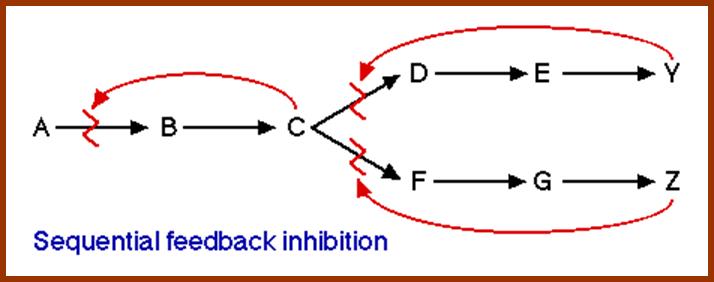
Common feedback inhibition mechanisms, (1) The basic feedback inhibition mechanism, where the product (P) inhibits the committed step (A->B). (2) Sequential feedback inhibition. The end products P1 and P2 inhibit the first committed step of their individual pathway (C->D or C->F). If both products are present in abundance, all pathways from C are blocked. This leads to a buildup of C, which in turn inhibits the first common committed step A->B. (3) Enzyme multiplicity. Each end product inhibits both the first individual committed step and one of the enzymes performing the first common committed step. (4) Concerted feedback inhibition. Each end product inhibits the first individual committed step. Together, they inhibit the first common committed step. (5) Cumulative feedback inhibition. Each end product inhibits the first individual committed step. Also, each end product partially inhibits the first common committed step.
http://www.edinformatics.com/
FACTORS
1. Temperature
Molecules of various dimensions are in constant motion at different rates within a living cytosolic fluid. When such moving substrates and enzymes of right kinds collide with each other, the enzymatic reaction is initiated. Thus the frequency of such collisions determines the rate of reaction. Higher the temperature, higher is the frequency of collision hence, the rate of biochemical reactions. Every enzyme has its own optimal temperature at which it shows the maximal activity. However, at very high temperatures, enzymatic proteins undergo denaturation, so the enzymatic activity is inhibited.
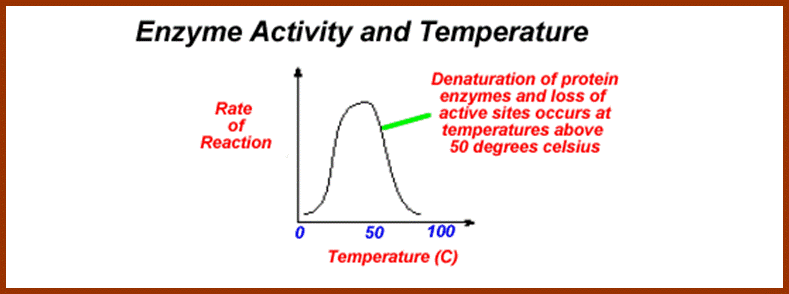
2. Hydrogen ion Concentration:
Intracellular ph has a profound effect on the total change of the enzyme. The structural integrity and functional capability of an enzyme depends upon its charges at specific active sites. As different enzymes contain different composition of amino acid residues, enzymes require a favorable pH for their activity. Many enzymes are active at 5.8-6.8 some have an optimal activity at acidic pH and some at alkaline ph. Thus the intracellular pH regulates enzyme mediated cellular activities.
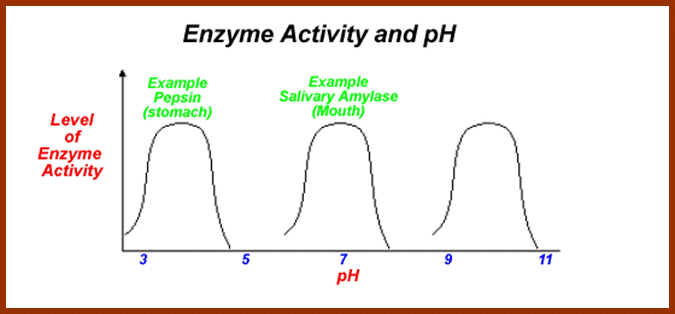
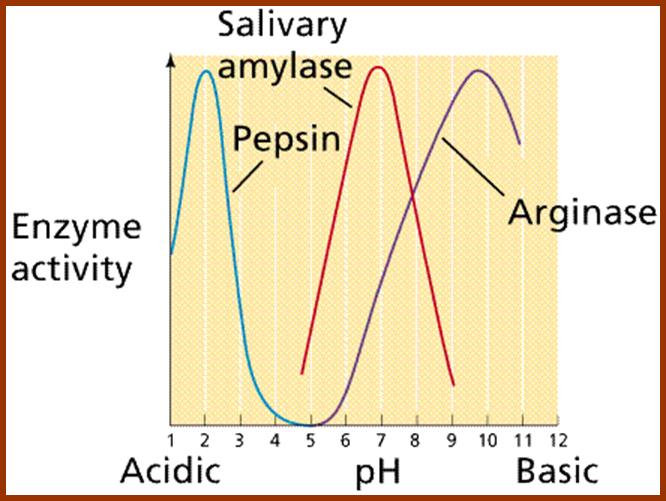
http://www2.estrellamountain.edu/
CONCENTRATION OF WATER
Water is a very important component of cellular protoplasm, in which all kinds of structures and inorganic ad organic substances are in suspended or in dissolved state. Water provides the medium for the molecules to move, collide and react. If the content of water is reduced, all the components get crowded in a given area and the enzymatic activity is reduced because of lack of opportunity for the right kind of collision between the substrate and enzymes. For example, dry seeds exhibit very low rate of respiration. But when dry seeds are provided with water, the cells absorb water and its content increases from 6% to 90%. With the increase in water content, respiratory activity also increases till the water content reaches saturation point.
CONCENTRATION OF SUBSTRATES AND ENZYMES
Enzymes, being functional molecules, require substrates for their further activity. If the concentration of substrates available is less than the number of enzymes present at any given time, only few enzymes are involved in enzyme reactions and the others remain idle. On the other hand, if the concentration of substrates is increased steadily, the number of enzymes reacting with substrates increases and thus the rate also increases. At a particular concentration or higher than that, the rate of activity remains constant because all the enzymes are loaded with substrates and maximum activity is brought about; hence the rate of enzymatic activity remains constant.
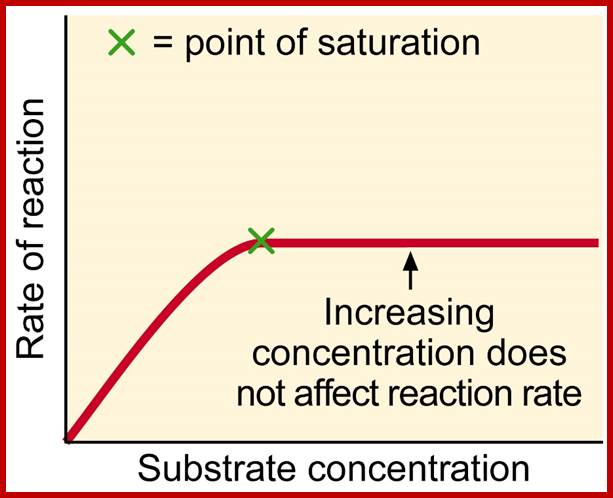
http://www.rsc.org/
On the contrary, the concentration of enzymes also has a say in the overall rate of biochemical activity. In a situation, where there is abundant supply of substrates, but the concentration of enzymes is very low. The rate achieved in such a situation is minimum, though individual enzymes show, maximum activity. If the concentration of enzymes is increased, the rate of reaction also increases tremendously. If the proportions of enzymes and substrates are equally raised, the rate of reaction steadily increases till other factors act as limiting factors.
OTHER FACTORS
Apart from the above said factors, the enzymatic activity also requires factors, like co-enzymes, co-factors, allosteric effectors, etc. for their optimal activity, particularly the availability of metallic ion sand organic co-enzymes are very important for the maximal activity of enzymes. Similarly certain regulatory factors also control the enzymatic activity without which no enzymatic activity is normal.
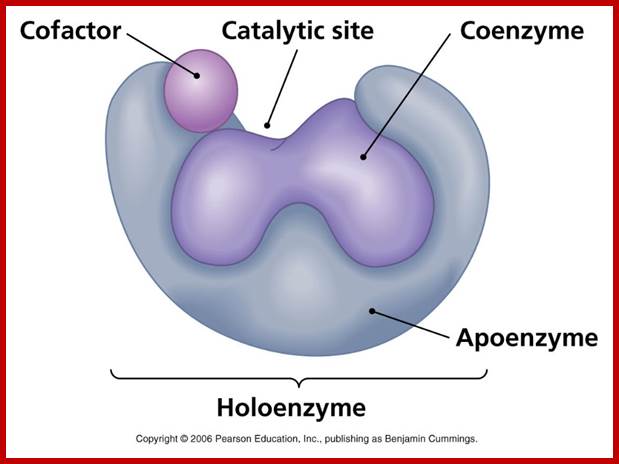
http://academic.pgcc.edu/
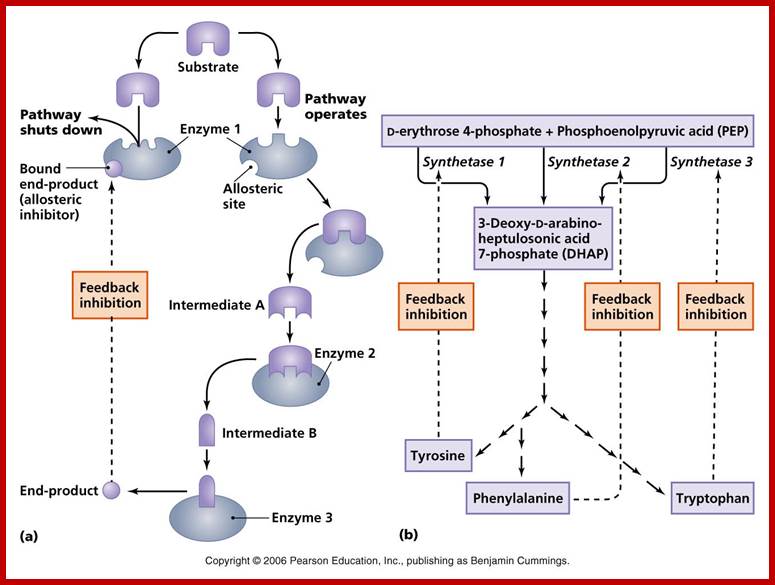
![]()
http://academic.pgcc.edu/
In allosteric inhibition (a), an enzyme having a secondary active site called an allosteric site close to the active site, preventing the formation of the enzyme-substrate complex.
In allosteric activation (b) the substrate binds to the allosteric site, allowing the active site to open. Allosteric enzymes can be used to regulate enzymatic pathways. In a negative-feedback pathway, the end product of a pathway binds to the allosteric site of the first enzyme, closing its active site and blocking the rest of the reactions.