In Vitro Mutagenesis:
Natural mutation, in general, is spontaneous. Any heritable change is considered as mutation. It can be spontaneous or induced. Mutation cannot be predicted in nature. Mutation at chromosomal level can be numerical (ploidy) or it can be structural (aberrations). At molecular level mutation can be deletion of a sequence of nucleotide or nucleotides, or addition or deletion of nucleotide or change in nucleotides’ structure, translocation of DNA segments with in the chromosome or between chromosomes or it can be due to inversion of DNA segments. Some DNA sequences rendered nonfunctional.
Point mutations are generally restricted to changes in a single nucleotide i.e. substitution of a nucleotide or addition of nucleotide or loss of a single nucleotide.
Mutation can lead to gain of a function called Gain mutation, or loss of a function called null mutation. Mutation can conditional, where the effect of mutation is expressed or manifest only under certain condition. Many mutations are spontaneously reversed called reverse mutations, but some mutations are suppressed or some suppress the function of other genes called suppressor mutations.
The rate of mutations and the kind of mutations vary from one to organism to the other, which depends upon the phylogenetic and hierarchical position of the organism. In that sense viruses show greater propensity for changes than Bacterial systems. And bacterial systems are more prone to mutations than higher organisms. Haploids suffer more than diploids due to mutation of one or the other kind. In diploids if there is one mutation in the homologous chromosomes is intact in diploids, it can be tolerated for one of the two (pair) remains of genes functional. One gene mutation an induce mutations in another gene.
All natural mutations are unpredictable and spontaneous and it is the primary force of nature that caused variations and those variations survived the test of the time are fixed and furthered, thus diversified species originated in nature. Plants can be used for mutations by drugs or radiations. Today molecular technology that is available at hand can be used create desirable mutation under in vitro condition. It is not just creating random mutations; it is now possible to create mutations to create new codons, new messages and new characters. This is possible to target a single nucleotide or a group of nucleotides. In this process one can change a single codon or add one or more codons and delete one or more codons. Such changes in coding sequence of a particular gene can generate a protein with new conformation or new function or both; in essential it amounts to protein engineering.
Protein engineering is an emerging technology of great importance in medicine and industry. By this technology one can enable the protein to be stable at higher temperature and still active in non-aqueous conditions. One can make the protein to change its affinity to the substrate and increase the activity by several folds at low Km. It is also possible to change its specificity of the substrate and function. This is going to be one of the futuristic aspects of biotechnology.
How proteins are engineered:
- Thermo stable T4 Lysosome: - Changing few amino acids to cysteine that produces more disulfide bonds make the proteins resistant to higher temperatures. Addition of cysteine at specific sites should not change the 3-D structure and the function of the protein, yet it is stable at high Tm.
- Thermo stable Triose phosphate isomerase: Increase in temperature leads to deamidation of Aspargine and Glutamine. This deforms the protein and so it looses its activity at higher Tm. But by in vitro site directed mutagenesis amino acid Aspargine at 14 and 78 can be changed to Threonine or Isoleucine. These changes have made the proteins to be thermo stable.
- Cloned b-interferon: Interferons produced in bacteria were not active because of dimerization and multimerization. The proteins were found to be inactive. By in vitro mutagenesis one or two cysteine were changed to serine. This prevents the individual subunits from dimerization or multimerization, yet they are found to be active.
- Active site modification: Tyrosyl tRNA synthase is the enzyme responsible for adding tyrosine to the tRNA. By changing the Threonine at 51-st position to Proline the enzyme has increased specific activity and increased specificity.
- Changed Staphylococcus nuclease (S1 nuclease); The S1 nuclease recognizes both ss RNA and ssDNA and dsDNA and acts at A.U or A.T rich regions and cuts to generate 3’phosphate and 5’OH groups. When Lysine at 116 positions was changed to cysteine, the protein showed remarkable site-specific activity.
- Subtisilin: It is a bacterial protein used in detergents. This protein is responsible for removing proteinaceous dirt. But the protein is susceptible for bleach and it is rendered inactive. By changing and addition of one or two cysteine residues, the number of S-S bonds increase. This had made the proteins to be stable not only in the detergent but also stable to bleach. Such engineered protein is used in detergent powder called ‘Biodetergent’.
In Vitro Random Mutation:
First method:
Using chemicals that act on DNA creates mutation, but the site of mutation is random. Whether or not ‘in vitro mutation’ has any desirable feature; it is perhaps a chance.
In order to induce mutations, obtain ssDNA from M13/18 plasmids with a gene of interest.
The ssDNA is subjected very low level of sodium bisulfate under controlled conditions such that the number of sites mutated will be very very minimal. Sodium bisulfate deaminates cytosine residues to uracil. When the DNA containing uracil replicates it produces DNA with Adenine in place of uracil. Thus whatever limited numbers of cytosine residues converted into adenines change the message, so also the protein product.
Such deaminated and oxygenated ssDNA is copied into dsDNA by using an universal primer. Such replicative form of DNA is used for bacterial transformation. Then the colonies are screened for any mutation. This way one can generate a large number of point mutation.
Second method:
Some of the E.coli strains such as Ung ( – )and Dut(+) are very useful in obtaining in vitro mutation, again it is random. Ung – strains are incapable of removing uracil nucleotides from the DNA by deglycosylation reaction. Dut – strains are lacking UTPase, thus the concentration of UTP inc5reases and UTPs are incorporated into the DNA. But Ung+ and Dut + strains have functional enzymes.
If a M13/18 plasmid containing a specific gene is used to transform double mutant strain, UTPs are incorporated into the replicating DNA and incorporated UTPs are not removed by N’deglycosylases, because of defective enzymes.
Obtain positive ssDNA from M13/18 transformed strains and using universal primers generate ds DNA.
Digest the plasmid in order to release the insert and clone the same into another plasmid and transform Ung + and Dut – bacterial strains. As the plasmid DNA replicates all the Us are removed by deglycosylases and the replicated plasmid is obtained.
Obtain the plasmids and release the insert and recline the gene and analyze for the random mutants.
Site directed Mutagenesis:
If the sequence of the desired DNA is known, from that one can find out certain restriction enzyme sites. For example within the gene of interest, assume there is one E.coR I site; the sequence of it is GAATTC.
If the site is cut with E.coR I enzyme it generates 5’ overhangs in both strands of the DNA.
----5’G AATTC---3’
----3’CTTAA G---5’
If such DNA with sticky ends is treated with S1 nuclease it removes the overhangs to blunt ends.
-----5’G C---3’
-----3’C G----5’
When such ends are ligated, one gets the DNA with 5’GC3’ sequences and four base pairs are lost. This leads not only to deletions but also changes the reading frame.
Similarly, if the cut ends are treated with T4 DNA polymerase with dNTPs, the enzyme fills the gaps of each ends by extending the cut ends by 5’à3’ direction to produce blunt ends. If such blunt ends are ligated, they generate 5’GAATTAATTC3’. In these four base pars are added. Again, this causes the addition and the reading frame is changed. Mostly this kind of changes doesn’t produce any functional protein, if by chance the changed reading frame works you are in luck.
Method II:
Site directed Mutagenesis”
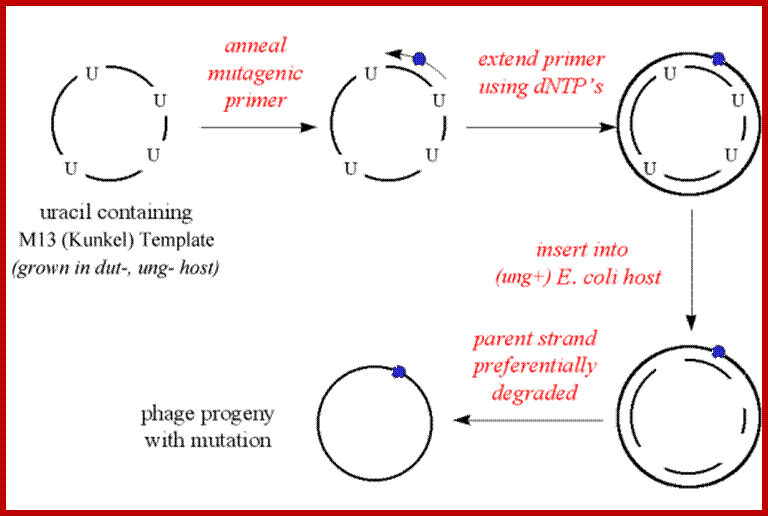
'Kunkel' method:
The M13mp vector with insert is first grown in a mutant E. coli host (e.g. CJ236), which would occasionally incorporate uracil into the DNA instead of thymidine.
- E. coli normally synthesizes an enzyme (uracil-N-glycosidase) that removes uracil residues in DNA. However, in ung- strains, the uracil is not removed
- The level of uracil mis-incorporation into DNA is enhanced in strains, which have a deficiency in dUTPase. This enzyme converts dUTP to dUDP, and therefore, in dut- strains the levels of dUTP are elevated and enhance misincorporation of dUTP into host DNA
- The E. coli strain CJ236 has a genotype which includes dut-/ung- features
- A mutagenic primer would be annealed to this single strand template (e.g. to produce a point mutation)
- The primer is extended using the four dNTP's, and is ligated to produce duplex DNA
- The duplex DNA is inserted into a different host E. coli (e.g. JM101) which recognizes and excises (degrades) uracil containing DNA (i.e. a strain which is ung+)
- The parent (wild type) strand is preferentially degraded and the mutagenic strand is replicated.
- The phage progeny typically have a high incidence (80-90%) of the desired mutation. ‘http://www.scrigroup.com/’
http://www.scrigroup.com/
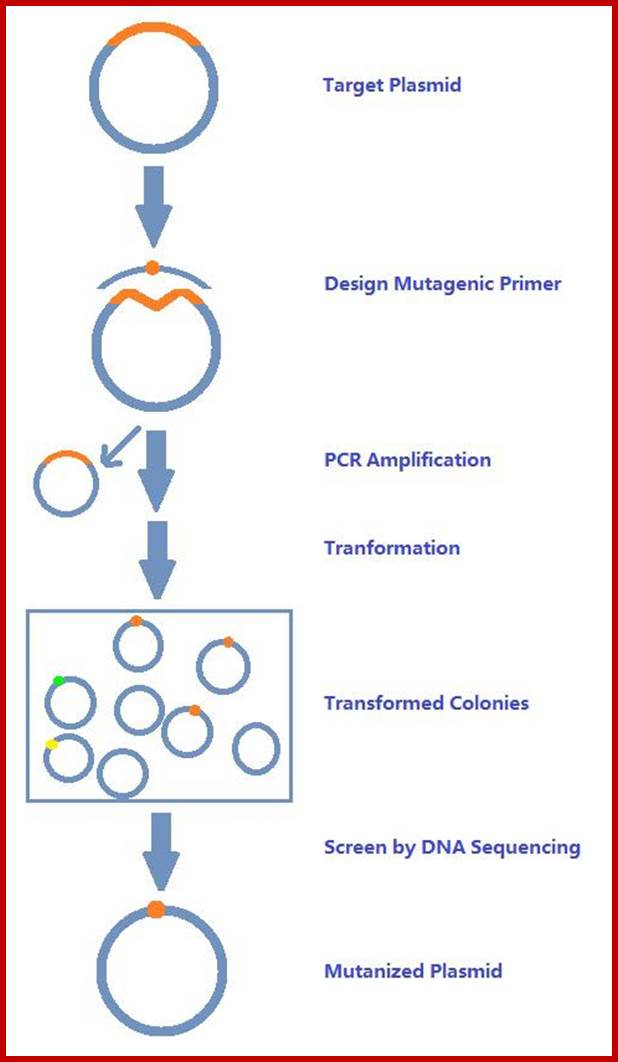
Site Directed Mutagenesis; http://www.creative-biogene.com/
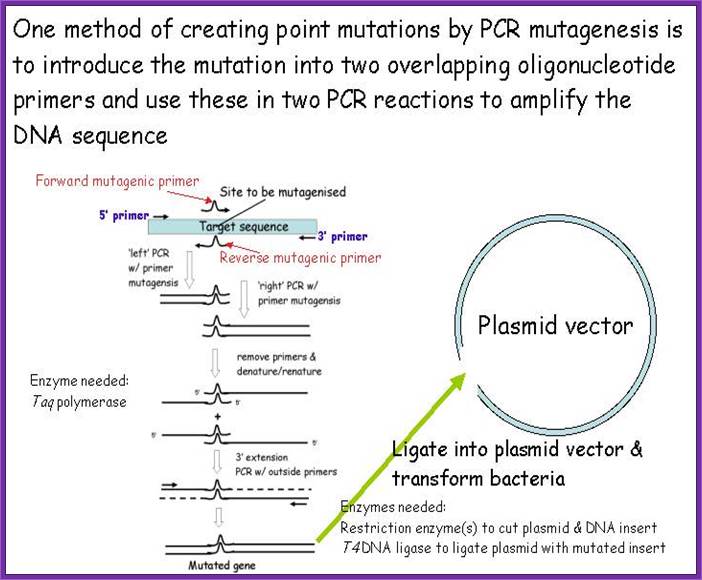
In this case the change is directed to one or more specific nucleotides thus one can change only one of the codons. For example, one wants to change Phenyl alanine (UUU) to Cysteine (UGU), design primers in such a way UUU is changed to UGU, where with exception of one nucleotide others are complementary to the nucleotides on either side of the said nucleotide.
5’--UUUà
3’ AAAà
Primer to the bottom strand- wit5h changed nucleotide-----5’UGUà
Primer to the top strand with changed nucleotide --- -<-ACA 5’
Melt the DNA to ssDNA and anneal the primer to the strands not to a state of stringency. This will provide an opportunity for the base pairs, which are not complementary, remain unpaired. Amplify the DNA using PCR protocols. Then melt the DNA to single strands and anneal them at high Tm for stringency.
Then use the plasmids and transform them and look for the change in the codon.
Another way is to have marker in the DNA which one wants to use for site directed mutation. Take a plasmid with the desired gene and a marker out side the gene such as a restriction site which can be cut with only Dpn-I. Use the primers and amplify the DNA. Then melt the DNA and Anneal. The parental strands anneal and new strands anneals by themselves. After annealing the DNA strands, cut the dsDNA with Dpn-I. The new strands have no Dpn-I site for the enzyme requires methylated sites, otherwise it does not recognize the site. Thus only the parental strands are cut, but not the new strands. When such DNA is used for bacterial transformation the cut DNA cannot transform and the uncut DNA transforms bacteria, thus one obtains the DNA with changed sequence to generate a new codon.
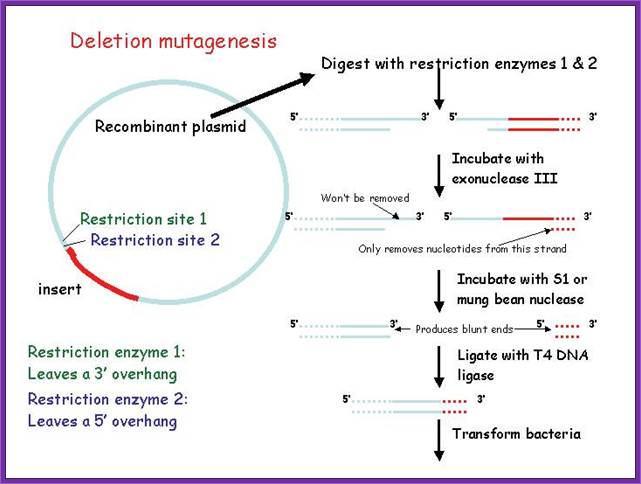
Deletion mutagenesis; Plant Cell Biol
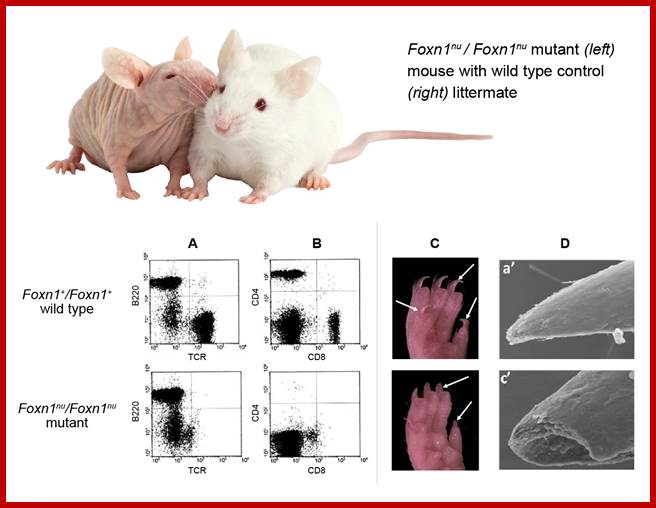
Figure 2 (click to zoom): Phenotypic
comparison of normal wild type and immunodeficient mutantFoxn1nu/Foxn1nu mouse strains. The
‘nude’ (Foxn1nu) mutation is a single base-pair (G) deletion
in exon 3, resulting in a frameshift mutation and termination at a TGA stop
codon in exon 6. Additional spontaneous and genetically engineered mouse Foxn1 mutations differing in their sequence
changes are known.
lower panel: FACS profiling of peripheral blood leukocytes shows absence of
T-lymphocyte markers in the mutant mouse when stained for (A) the T-cell receptor (TCR) and
B-lymphoid marker, B220 or (B)T-cell
activation co-receptors CD4 and CD8.
Nail morphology in wild-type and mutant mice.
Shown are (C) hind-paw photos and(D) scanning EM of nails. Nail plates of
normal adults are smooth, hard, and have sharp tips while mutant nails are
broken and have blunt ends.
http://www.informatics.jax.org/
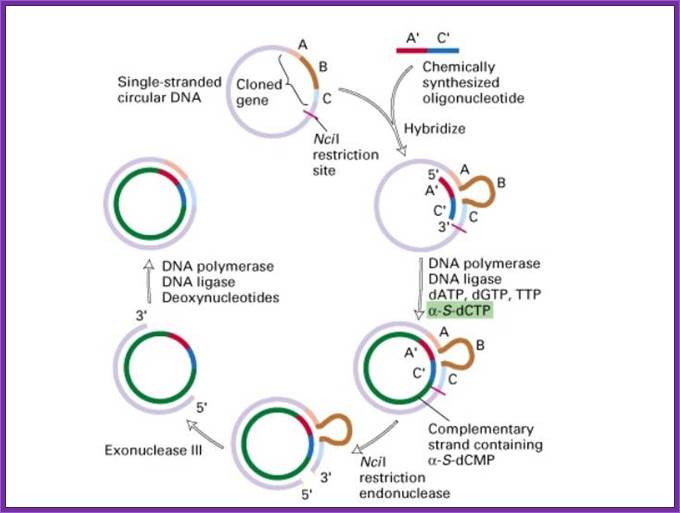
Oligo based mutagenesis; http://www.tankonyvtar.hu/
-0-0-
Application of Site Directed Mutagenesis:
· Proteins stable and functional at high temperatures.
· Proteins stable and functional at changed pH.
· Proteins active in non-aqueous solvents.
· Proteins don’t require cofactors for their function.
· Proteins resistance to proteases.
· Proteins with changed allosteric features.
· Proteins with changed active site and increased specific activity.
· Proteins with changed Km and Vmax with increased catalytic efficiency.
A list of few engineered proteins:
· Alpha Amylase- used in bear making.
· Subtisilin-bio-detergent.
· Amino cyclase- Preparation of L-amino acids.
· Bromelain- Meat tenderizer and juice clarifier.
· Catalase- anti oxidant in food preparation.
· Ficin- meat tenderizer.
· Cellulase- alcohol and glucose production.
· Gluco amylase- beer making and other EtOH products.
· Glucose oxidase- anti oxidant in food processing.
· Invertase- sucrose inversion.
· Lipase- cheese making and flavoring.
· Papain- meat tenderizer and juice clarifier.
· Pectinase- juice clarifier.
· Protease- detergent preparation.
· Rennet- cheese making.
· Now it can be more than thousand,