PCR Techniques:
Polymerase chain reaction is techniques developed in 1980s by Karry Banks Mullis, Nobel Laureate 1993. It has been a hit since then. Basic premise of these techniques is to amplify a DNA piece to the required quantity from a source as scarce as few Pico moles. This is an incredible, yet a simple technique procedure wise. All that is required is the knowledge of sequence of the DNA or the sequence at its borders. Using the knowledge of the sequence one can design primers to both strands in opposite orientation, so that both strands are amplified in synthesis.
5’GATC------------------------------------TATA3’
3’CTAG------------------------------------ATAT5’
The middle region (blue is the target DNA to be amplified. The border pink sequence is used for generating primers.
The primers for the top strand will be 5’TATA and the primers for the bottom strand will be 5’GATC.
When such double stranded DNA segment is heated to 95oC both strands melt and separate. If one adds two primers developed and brought the temperature down to 55^oC the primers anneal to their sequences. When sufficient amount of dNTPs and heat resistant enzyme like Taq polymerase, usually one unit of enzyme per vial of 50ul reaction mix, is added and the temperature is raised to 72^oC, which is the optimum Tm for the enzyme, it uses the 3’ ends of the primers and extends the primer till it reaches their respective ends of the DNA fragment, so on both strands a new complementary strands form. Thus a single dsDNA is duplicated into two dsDNA. If the protocol is repeated once again, i.e. heat to 94^oC and hold it for one minute to melt the dsDNA into ssDNA, bring the temperature to 55 ^oC and hold it for a minute or two for annealing of primers. Then increase the temperature to 74 degrees and hold it for 2 minutes for the extension of the primer into complementary strands; now two ds DNAs have become four dsDNA. If this protocol is repeated 32 times a single dsDNA will be amplified into 107, 374, 1824 ds DNA molecules. So the amount of DNA at the source can be as small as one molecule (theoretically). If a small speck of blood or one hair follicle is enough to obtain sufficient amount of DNA for PCR amplification. However the enzyme has low processivity and low fidelity.
In recent time proof reading enzymes have been obtained; they are Taq polymerases from Perkin Elmer and ULTMA from Stratagene (?). They are believed to have good processivity and reasonably good fidelity. Another feature is efficiency of amplification of longer DNA more than 3-10kbp. In such cases one has to optimize the concentration Mg2+ ions. Some people are using what is called Hot-start. It means melt the DNA then bring to annealing temperature and then raise the Tm for primers extension; at that point of time the enzyme is added.

Eppendorf Mastercycler nexus Family;
http://www.pocdscientific.com.au/
10x PCR buffer:
670 mM Tris Cl pH 9.0,
67 mM MgCl 2,
1.7 mg per ml BSA,
166 mM (NH4) 2 SO4.
The total reaction mix is 25 ul or 50 ul all included. Over the mix a drop of mineral oil is added in order to prevent loss of liquid by evaporation.
The enzyme is heat resistant enzyme. These are isolated from organisms grow in the vicinity of hot springs. These organisms grow in such hostile and hot environment; the enzymes in such organisms are capable withstanding such high temperatures of 94^oC for 60 to 90 minutes. The study of amino acid sequence and the way these proteins are folded into 3-D structure is a paradigm for genetic engineering of proteins to be stable at high temperatures.
These reactions are automated and depending upon the annealing Tm one has to program the Tm. Once the program is fed into the machine it will take care of the process. At the end of set number of cycles one has keep the mix in elongation mode for 10-15 minutes, some of the DNA ends which are not complete will be extended to completion. Interesting feature polymerase amplification is that the amplified DNA fragments will have A as an n overhang. As the amplified DNA is phosphorylated at the 5’ end, after purification of PCR products, they can be used for direct ligation to vectors, which have a T as an overhang.
DNA
With Reaction mixture
![]()
94 ^o- 2minutes
55^oC – 2minutes
74^oC- for 2 minutes
![]() 94^oC
for 2 minutes
94^oC
for 2 minutes
![]() Number
of cycles-
Number
of cycles-
Last cycle 10-15 minutes at 74^oC
![]()
Freeze till it is used.
Then the sample is analyzed on an Agarose gel.

Some of the 32 (known?) PCR techniques:
Standard PCR:
In the few years since its introduction, the polymerase chain reaction has become a widespread research technique. This popularity of the PCR is primarily due to its apparent simplicity and high probability of success. In fact, the PCR is a relatively complicated and, as yet, incompletely understood biochemical brew; where constantly changing kinetic interactions among the several components determine the quality of the products obtained. Although good results will be obtained in most cases, there are a number of parameters that can be explored if better results are required or if the reaction fails altogether.
Because of the great variety of applications in which PCR is used, it is probably impossible to describe a single set of conditions that will guarantee success in all situations. Nevertheless, the reaction outlined below proved to be adequate for most amplifications and in those cases where problems are encountered, it provides at least a starting point from which modifications can be attempted (e.g., the Mg2+ concentration should be checked in every new PCR setup).
The standard PCR is typically done in a 50 μl volume
and, in addition to the sample DNA, contains 50mM KCl, 10mM Tris ![]() HCl (pH 8.4), 1.5mM MgCl
HCl (pH 8.4), 1.5mM MgCl ![]() , 100
, 100 ![]() g/ml gelatin, 0.25
g/ml gelatin, 0.25 ![]() M of each primer, 200
M of each primer, 200 ![]() M of each deoxynucleotide triphosphate (dATP, dCTP, dGTP, and dTTP), and 2.5 units of Taq
polymerase . The type of the DNA sample will be variable,
but it will usually have between 10
M of each deoxynucleotide triphosphate (dATP, dCTP, dGTP, and dTTP), and 2.5 units of Taq
polymerase . The type of the DNA sample will be variable,
but it will usually have between 10 ![]() and 10
and 10 ![]() copies of template (e.g., 0.1
copies of template (e.g., 0.1 ![]() g human genomic DNA).
g human genomic DNA).
Allele specific PCR;
High-Throughput SNP Genotyping by Allele-Specific PCR with Universal Energy-Transfer-Labeled Primers
1. Maxim V. Myakishev1,3, Yuri Khripin2, Stella Hu1, and Dean H. Hamer1
Abstract
We have developed a new method for high-throughput genotyping of single nucleotide polymorphisms (SNPs). The technique involves PCR amplification of genomic DNA with two tailed allele-specific primers that introduce priming sites for universal energy-transfer-labeled primers. The output of red and green light is conveniently scored using a fluorescence plate reader. The new method, which was validated on nine model SNPs, is well suited for high-throughput, automated genotyping because it requires only one reaction per SNP, it is performed in a single tube with no post-PCR handling, the same energy-transfer-labeled primers are used for all analyses, and the instrumentation is inexpensive. Possible applications include multiple-candidate gene analysis, genome wide scans, and medical diagnostics.
A molecular method for S-allele identification in apple based on allele-specific PCR. G. A. Janssens, I. J. Goderis, W. F. Broekaert and W. Broothaerts
cDNA sequences
corresponding to two self-incompatibility alleles (S-alleles) of the
apple cv ![]() Golden
Delicious
Golden
Delicious![]() have
previously been described, and now we report the identification of three
additional S-allele cDNAs of apple, one of which was isolated from a
pistil cDNA library of cv
have
previously been described, and now we report the identification of three
additional S-allele cDNAs of apple, one of which was isolated from a
pistil cDNA library of cv ![]() Idared
Idared![]() and
two of which were obtained by reverse transcription-PCR (RT-PCR) on pistil RNA
of cv
and
two of which were obtained by reverse transcription-PCR (RT-PCR) on pistil RNA
of cv ![]() Queen's
Cox
Queen's
Cox![]() . A comparison
of the deduced amino acid sequences of these five S-allele cDNAs
revealed an average homology of 69%. Based on the nucleotide sequences of these
S-allele cDNAs, we developed a molecular technique for the diagnostic
identification of the five different S-alleles in apple cultivars. The
method used consists of allele-specific PCR amplification of genomic DNA
followed by digestion of the amplification product with an allele-specific
restriction endonuclease. Analysis of a number of apple cultivars with known S-phenotype
consistently showed coincidence of phenotypic and direct molecular data of the S-allele
constitution of the cultivars. It is concluded that the S-allele
identification approach reported here provides a rapid and useful method to
determine the S-genotype of apple cultivars.
. A comparison
of the deduced amino acid sequences of these five S-allele cDNAs
revealed an average homology of 69%. Based on the nucleotide sequences of these
S-allele cDNAs, we developed a molecular technique for the diagnostic
identification of the five different S-alleles in apple cultivars. The
method used consists of allele-specific PCR amplification of genomic DNA
followed by digestion of the amplification product with an allele-specific
restriction endonuclease. Analysis of a number of apple cultivars with known S-phenotype
consistently showed coincidence of phenotypic and direct molecular data of the S-allele
constitution of the cultivars. It is concluded that the S-allele
identification approach reported here provides a rapid and useful method to
determine the S-genotype of apple cultivars.
Enhanced allele-specific PCR discrimination in SNP genotyping using 3′ locked nucleic acid (LNA) primers†
- David Latorra, Krista Campbell, Andreas Wolter, J. Michael Hurley
The specificity and reliability of locked nucleic acid (LNA) substitution at the 3′ position of allele-specific PCR (AS-PCR) primers for SNP detection was investigated in direct comparison to DNA primers. Both plasmid and human genomic DNA templates were examined in this study. All possible DNA and 3′ LNA mismatch combinations were tested in triplicate with the plasmid target. LNA primers yield consistently low amounts of mismatch products with all base combinations, whereas certain mismatches with DNA primers generate strong false positive amplicons. Amplified human SNP alleles within the cystic fibrosis (CFTR) gene were analyzed in AS-PCR by gel analysis and real-time fluorescence generation. A 3′ LNA residue in the primer at the SNP site improves allelic discrimination and functions under a wide window of PCR conditions. We demonstrate increased AS-PCR specificity with comparable sensitivity using 3′ LNA primers in gel electrophoresis and real-time detection experiments. This increase in AS-PCR discrimination with 3′ LNA primers should facilitate the use of this simple, rapid, and inexpensive technique for SNP genotyping applications. Hum Mutat 22:79–85, 2003.© 2003 Wiley-Liss, Inc.
Suppression Subtractive Hybridization (SSH):
Subtractive hybridization is an approach allowing comparison of two DNA populations and isolation of fraction enriched in differentially distributed molecules. Subtractive hybridization is usually applied to identify genes with differential expression pattern, in particular genes involved in the regulation of basic biologicla processes.
Suppression Subtractive Hybridization (SSH) is the most famous of subtraction methods (Lukyanov et al., 1994; Gurskaya et al., 1996; Diachenko et al., 1996). It based on the PCR suppression by inverted terminal repeats (PS-effect). Upon initiation of PCR after the denaturation phase, the single-stranded (ss) DNA fragments flanked by inverted terminal repeats (ITR) may form either self-annealing "pan-like" structures (preventing the primer binding to its complementary binding sites and suppressing the PCR) or DNA/primer hybrid structures. In the latter case, if the primer corresponding to the outer part of ITR is used, the DNA synthesis by Taq-polymerase restores the original structure, ensuring the persistence of suppression during further PCR cycles. In complex mixtures, the use of PCR suppression allows selective amplification of molecules that are flanked by different adapters at opposing termini (asymmetrically flanked molecules).

Schematic outline of PCR suppression by inverted terminal repeats; http://www.evrogen.com/
Touchdown polymerase chain reaction or touchdown style polymerase chain reaction Wiki- It is a method of polymerase chain reaction by which primers will avoid amplifying nonspecific sequences. The annealing temperature during a polymerase chain reaction determines the specificity of primer annealing. The melting point of the primer sets the upper limit on annealing temperature. At temperatures just below this point, only very specific base pairing between the primer and the template will occur. At lower temperatures, the primers bind less specifically. Nonspecific primer binding obscures polymerase chain reaction results, as the nonspecific sequences to which primers anneal in early steps of amplification will "swamp out" any specific sequences because of the exponential nature of polymerase amplification.
The earliest steps of a touchdown polymerase chain reaction cycle have high annealing temperatures. The annealing temperature is decreased in increments for every subsequent set of cycles (the number of individual cycles and increments of temperature decrease is chosen by the experimenter). The primer will anneal at the highest temperature which is least-permissive of nonspecific binding that it is able to tolerate. Thus, the first sequence amplified is the one between the regions of greatest primer specificity; it is most likely that this is the sequence of interest. These fragments will be further amplified during subsequent rounds at lower temperatures, and will out compete the nonspecific sequences to which the primers may bind at those lower temperatures. If the primer initially (during the higher-temperature phases) binds to the sequence of interest, subsequent rounds of polymerase chain reaction can be performed upon the product to further amplify those fragments.
PCR-Restriction Fragment Length Polymorphism Method for Detection of Cyclospora cayetanensis in Environmental Waters without Microscopic Confirmation;
1. Joan M. Shields and
We developed an alternative nested-PCR-restriction fragment length polymorphism (RFLP) protocol for the detection of Cyclospora cayetanensis in environmental samples that obviates the need for microscopic examination. The RFLP method, with the restriction enzyme AluI, differentiates the amplified target sequence from C. cayetanensis from those that may cross-react. This new protocol was used to reexamine a subset (121 of 180) of surface water samples. Samples previously positive when the CYCF3E and CYCR4B primers (33) and RFLP with MnlI (20) were used were also PCR positive with the new primers; however, they were RFLP negative. We verified, by sequencing these amplicons, that while two were most likely other Cyclospora species, they were not C. cayetanensis. We can detect as few as one oocyst seeded into an autoclaved pellet flocculated from 10 liters of surface water. This new protocol should be of great use for environmental microbiologists and public health laboratories
Single cell PCR
Single cell PCR has applications in many areas, and has great application especially in the field of prenatal diagnostics. In prenatal diagnosis, single cell PCR has made possible preimplantation genetic analysis and the use of fetal cells enriched from the blood of pregnant women for the assessment of single-gene Mendelian disorders. Single-cell PCR has not only proven its usefulness in diagnostics, but also lately has been very useful to basic scientists investigating immunological, neurological and developmental problems.
Recent advances in molecular biology techniques such as whole genome amplification (WGA) procedures and single-cell complementary DNA arrays will permit the genetic analysis of single cells to become a more common practice, thus creating new avenues for diagnosis and research.
Single-cell
molecular-biology is a relatively new scientific branch in biology. The first
single-cell analysis were involved in the characterization of mitochondrial DNA
in 1988. Single-cell DNA analysis, in particular genomic DNA, is important and
may be informative in the analysis of genetics of cell clonality, genetic
anticipation and single-cell DNA polymorphisms. Nowadays for most scientists
the quantitative transcriptomics in a single-cell is much more
important, and the analytical method of choice is the quantitative real-time
RT-PCR. The relative abundance of single mRNAs and
their up- or down-regulation in a single cell, compared to their neighbour
cells, is the goal. The need for quantitative single-cell mRNA analysis is
evident given the vast cellular heterogeneity of all tissue cells and the
inability of conventional RNA methods, like northern blotting, RNAse protection
assay or classical block RT-PCR, to distinguish individual cellular
contributions to mRNA abundance differences.
The purpose of this single-cell PCR page is
to provide researchers with resources (papers, talks, posters) for single-cell
molecular analysis such as RT-PCR and quantitative PCR:
Alu PCR,
·
Definition
PCR using
a primer that anneals to Alu repeats to amplify DNA located between two
oppositely oriented Alu sequences. Used as a method of obtaining a fingerprint
of bands from an uncharacterized human DNA.
Alu PCR is a rapid and easy-to-perform "DNA fingerprinting" technique based on the simultaneous analysis of many genomic loci flanked by Alu repetitive elements, which allows the detection of genetic polymorphisms and mutations in human and primate genomes. In the protocol described in the present chapter, two fluorochrome-labelled primers complementary to Alu sequences are used to perform the PCR, and the amplification products are then analysed by capillary electrophoresis. The resulting -complex electrophoretic pattern may show sample-to-sample variability due to insertion, deletion, or sequence change of Alu retrotransposons, or caused by length variation of the sequence interposed between two Alus (Medline)

Human DNA typing by the
Molecular Biology Lab:
Determination of an ALU insertion polymorphism by PCR: Biology in the genomic
Ages: https://www.amherst.edu/
![]()
What is asymmetric PCR? A PCR in which the predominant product is a single-stranded DNA, as a result of unequal primer concentrations. As asymmetric PCR proceeds, the lower concentration primer is quantitatively incorporated into double-stranded DNA. The higher concentration primer continues to primer synthesis, but only of its strand.
This protocol describes how to carry out asymmetric PCR and single-sided PCR. Both types of PCR are used to produce single-stranded DNA. Asymmetric PCR initially generates a substantial amount of double-stranded DNA and the double-stranded DNA is then used to generate single-stranded DNA. This approach is especially useful for direct sequencing of PCR products. Single-sided PCR uses only a single primer to generate single-stranded fragments either for sequencing purposes or for use as single-stranded probes. Single-sided PCR is carried out in the same manner as ordinary PCR except that one of the primers is left out of the reaction.
Colony PCR,
Colony PCR can be used after a transformation to screen colonies for the desired plasmid. Primers are used which generate a PCR product of known size. Thus, any colonies which give rise to an amplification product of the expected size are likely to contain the correct DNA sequence.
Colony PCR is now a widely-used method for quickly screening large numbers of bacterial cells for a gene of interest. When screening recombinant clones, PCR screening of colonies decreases the screening time by one full day compared with miniprep-based methods. Use of a hot-start polymerase in the PCR also allows the convenience of leaving the PCR reactions at room temperature for up to 24 hours before processing, something that becomes attractive when you have a large number of colonies to process or a long walk to fill your ice bucket.
Quantitative Competitive PCR;
Quantitation of transcription via RT-PCR can be approached using either relative RT-PCR or a competitive RT-PCR strategy. Theoretically, relative quantitation by RT-PCR can grossly estimate differences in abundance of a particular transcript between samples if the following conditions are met:
1. The amount of RNA used in the initial cDNA synthesis reaction is precisely
controlled.
2. The amount of cDNA used in the PCR reaction is precisely controlled.
3. The number of PCR cycles necessary to generate enough product to detect is
not past the exponential phase of the PCR.
In practice, it is very difficult to ensure that these criteria are met. Of these conditions, only the amount of input RNA must be strictly controlled to use competitive RT-PCR. The validity of the competitive amplification approach has been confirmed in numerous studies and it is the basis for several quantitative PCR and RT-PCR assays designed to quantitate human pathogens.
Development of a Quantitative Competitive PCR Assay for Detection and Quantification of Escherichia coliO157:
QC-PCR has been used to detect and determine bacterium numbers for a variety of difficult-to-culture bacteria. The method is based on the co- amplification of the sequence to be quantified (the target sequence) with a known amount of another sequence (the competitor) which resembles the target. Both sequences amplify with the same primers. These two sequences should ideally be from the same region of DNA in order for the primers to amplify each with equal efficiency but should differ slightly in size to be distinguished by agarose gel electrophoresis. For QC-PCR, a dilution series of three to five PCR reaction mixtures are made, each with a constant (unknown) amount of added target DNA and a known dilution series of competitor DNA. The target and competitor DNA compete for the same primers; when the concentration of each is equivalent, band intensities will be equivalent. The point of equivalence is determined by visual assessment of band intensities or by digital analysis of the gel image and generation of a regression line (10). Quantitation of the gene copy number can be converted to chromosomal equivalents and cell numbers. The objectives of this study were to determine if QC-PCR could be applied to foods and to develop a quantitative PCR assay for detection and enumeration of E. coli O157:H7 cells in broth and skim milk.
Differential Display RT-PCR.
Differential-Display Reverse Transcription-PCR to Molecular Pathogenesis and Medical Mycology.
The host-fungus interaction is characterized by changes in gene expression in both host and pathogen. Differential-display reverse transcription PCR (DDRT-PCR) is a PCR-based method that allows extensive analysis of gene expression among several cell populations. Several limitations and drawbacks to this procedure have now been addressed, including the large number of false-positive results and the difficulty in confirming differential expression. Modifications that simplify the reaction time, allow the use of minute quantities of RNA, or address unusual species- or gene-specific sequences have been reported. DDRT-PCR has been used to address biological questions in mammalian systems, including cell differentiation, cell activation, cell stress, and identification of drug targets. In microbial pathogenesis and plant pathogenesis, DDRT-PCR has allowed the identification of virulence factors, genes involved in cell death, and signaling genes. In Candida albicans, DDRT-PCR studies identified TIF-2, which may play a role in the upregulation of phospholipases, and the stress-related genes, CIP1 and CIP2. In Histoplasma capsulatum and C. albicans, genes involved in the host-pathogen interaction, including a member of the 100-kDa family in Histoplasma and an ALS and 14-3-3 gene in Candida, were potentially identified by DDRT-PCR. Although very few reports have been published in medical mycology, studies in mammalian, nonfungal microbial, and plant pathogen systems are easily applied to basic questions in fungal pathogenesis and antifungal therapeutics.
Hot start PCR,
Hot-start" PCR is a modification of conventional PCR that reduces non-specific product amplification. In this procedure amplification cannot occur until the reaction temperature is above that where non-specific annealing of primers to targets occurs. This block in amplification is usually accomplished by using a DNA polymerase that is inactive until higher temperatures are reached.
Hot start” is a term used to describe the inactivation of a DNA polymerase until the initial denaturation step of PCR cycling. Hot start eliminates spurious amplification products resulting from non-specific priming events during reaction setup and initiation, and increases overall reaction efficiency. There are several methods used to inactivate DNA polymerases that include chemical modification (e.g. anhydrides or formaldehydes), physical modification (e.g. wax beads), aptamer binding, primer sequestration, antibody binding, and the addition of thermo-labile blocking groups on dNTPs or primers.
Immuno-PCR on chip
However, most natural processes involve proteins and other non-nucleic acids, so the analysis of nucleic acids is not sufficient to explore biological principles. To extend the scope of PCR to the high-sensitivity detection of proteins, Sano et al. established the immuno-PCR (IPCR) method [2]. Taking advantage of specific conjugates comprising an antibody and a DNA marker fragment, IPCR combines the versatility of enzyme-linked immunosorbent assays (ELISAs) with the amplification power and sensitivity of the PCR. As a consequence, the limit of detection (LOD) of a given ELISA is, in general, enhanced 100–10 000 fold by the use of PCR as a signal amplification system. IPCR has been refined from a research method to a well established technique for routine applications in both fundamental and applied immunological research. Applications of IPCR in various field of biomedical research have been reported.
However, most natural processes involve proteins and other non-nucleic acids, so the analysis of nucleic acids is not sufficient to explore biological principles. To extend the scope of PCR to the high-sensitivity detection of proteins, Sano et al. established the immuno-PCR (IPCR) method. Taking advantage of specific conjugates comprising an antibody and a DNA marker fragment, IPCR combines the versatility of enzyme-linked immunosorbent assays (ELISAs) with the amplification power and sensitivity of the PCR. As a consequence, the limit of detection (LOD) of a given ELISA is, in general, enhanced 100–10 000 fold by the use of PCR as a signal amplification system (Figure 6). IPCR has been refined from a research method to a well established technique for routine applications in both fundamental and applied immunological research. Applications of IPCR in various field of biomedical research have been reported.
In Situ PCR.
Definition of In Situ PCR
In Situ PCR (ISH) is a polymerase chain reaction that actually takes place inside the cell on a slide. In situ PCR amplification can be performed on fixed tissue or cells.

Flow chart describing the application of in situ RT-PCR to tissue microarrays. Step 1: Slide preparation. Sections were deparaffinated by xylene washes, and celluar material permeabilised by limited proteinase K digestion; Step 2: amplification of reverse-transcribed cDNA using intron-spanning PCR primers (horizontal arrows) added in a single mix of reverse transcriptase, rTth polymerase and deoxyribonucleotides spiked with digoxygenin-labelled dUTP (black ovals); Step 3: visualisation of PCR products by binding to digoxygenin-specific gold-labelled antibodies (yellow dots), followed by silver nucleation around the bound gold particles (grey crescents); http://www.jnanobiotechnology.com/
In situ hybridization (ISH) applies the methodology of the nucleic acid hybridization technique to the cellular level. Combining cytochemistry and immunocytochemistry, In Situ PCR allows the identification of cellular markers to be identified, and further permits the localization of to cell specific sequences within cell populations, such as tissues and blood samples.
In Situ PCR is limited to the detection of non-genomic material such as RNA, genes or genomes, as the detection limit in most conditions is several copies of the target nucleic acid per cell. Therefore, due to copy number limitations, hybridization of RNA is more sensitive than DNA detection.
Inverse PCR,
Inverse polymerase chain reaction (Inverse PCR) is a variant of the polymerase chain reaction that is used to amplify DNA with only one known sequence. One limitation of conventional PCR is that it requires primers complementary to both termini of the target DNA, but this method allows PCR to be carried out even if only one sequence is available from which primers may be designed.
Inverse PCR is especially useful for the determination of insert locations. For example, various retroviruses and transposons randomly integrate into genomic DNA. To identify the sites where they have entered, the known, "internal" viral or transposon sequences can be used to design primers that will amplify a small portion of the flanking, "external" genomic DNA. The amplified product can then be sequenced and compared with DNA databases to locate the sequence which has been disrupted.
The inverse PCR method involves a series of restriction digests and ligation, resulting in a looped fragment that can be primed for PCR from a single section of known sequence. Then, like other polymerase chain reaction processes, the DNA is amplified by the temperature-sensitive DNA polymerase:
- A target region with an internal section of known sequence and unknown flanking regions is identified
- Genomic DNA is digested into fragments of a few kilobases by a usually low-moderate frequency (6-8 base) cutting restriction enzyme.
- Under low DNA concentrations, self-ligation is induced to give a circular DNA product.
- PCR is carried out as usual, with primers complementary to sections of the known internal sequence.*
Finally the sequence is compared with the sequence available in the data base.
Note: although the figure suggests that the circularized ligation product is digested prior to PCR, this is not the case. PCR does not require linear products and the use of another restriction enzyme to cut the known sequence could also cut within the unknown region, resulting in a failed PCR (from WIKI).

Summary of the inverse PCR process; http://en.wikipedia.org/
ISSR PCR Inter sequence Specific PCR;
ISSR-PCR technique: a useful method for characterizing new allotetraploid somatic hybrids of mandarin.
Principle:
The simple sequence repeats or microsatellites especially (TAA)n and (AT)n repeats are ubiquitous in the silkworm genome. The changes in the number of repeats in a microsatellite array, which is considered to be the result of slippage of the DNA polymerase which occurs during DNA replication, provide an unlimited source of polymorphism. However, studying SSR is quite labor intensive since complete sequence information flanking the repeats is necessary to design primers for PCR amplification. Here, we demonstrate an alternative, efficient method that does not require any prior sequence information. In this method microsatellite sequence anchored either at 5' or 3' ends with a stretch of degenerate nucleotides were used for inter-repeat region amplification.
ADVANTAGES
- This method requires small quantity of DNA (5-20 ng/reaction depending up on the type of electrophoretic assay) and it is PCR based.
- This method provides dominant, reproducible and large number of markers.
- The whole analysis is automated to enable the researchers to carry out large-scale genetic mapping and population studies.
- This method overcomes the limitation of flanking sequence characterization as required by SSRs.

ISSR; http://www.ncbi.nlm.nih.gov/
STS polymorphisms that are found between microsatellite repeats. Primers can be designed based on a microsattellite reapeats exclusively, in which case this technique will target multiple loci due to known abundance of repeat sequences in the genome. Alternatively, primers can be extended outside or inside the ISSR in which case a unique region most likely will be amplified.

http://www.ncbi.nlm.nih.gov/
LIMITATIONS:
It is a dominant marker and hence not as informative as SSRs. ISSR-PCR. A single primer targeting a (CA)n repeat, anchored either at the 3' (green arrows) or at the 5' end (yellow arrows) of the repeat, is used to amplify genomic sequence flanked by two inversely oriented (CA)n repeat sequence.
Inter Simple Sequence Repeat region can be amplified using different protocols as per the lab’s requirement. In the past years we have tested many different protocols and standardized them for our population studies, polymorphism estimations, strain/variety identification, Mapping etc. Here, we list the best of our methods, which are tried and tested, in our lab for your use.
· Late PCR, Linear-After-The-Exponential (LATE)-PCR
Traditional asymmetric PCR uses conventional PCR primers at unequal concentrations to generate single-stranded DNA. This method, however, is difficult to optimize, often inefficient, and tends to promote nonspecific amplification. An alternative approach, Linear-After-The-Exponential (LATE)-PCR, solves these problems by using primer pairs deliberately designed for use at unequal concentrations.
LATE-PCR establishes straightforward primer design criteria for generating high yields of single-stranded DNA products without extensive reaction optimization. LATE-PCR effectively eliminates the need for multiple rounds of amplification or laborious steps for purifying single-stranded DNA. When combined with real-time or end-point detection using low-Tm probes, this method offers unique potential in forensics, bioweapons detection, and other fields where assay sensitivity and consistency are essential.
Ligation-anchored PCR: a simple amplification technique with single-sided specificity
1. A B Troutt, M G McHeyzer-Williams, B Pulendran, and G J Nossal
A simple, efficient, and sensitive technique has been developed for amplification of cDNAs encoding molecules with 5' regions of unknown sequence. In this ligation-anchored PCR, T4 RNA ligase is used to covalently link an "anchor" oligonucleotide to first-strand cDNAs. These anchored cDNAs are then amplified by using one PCR primer specific for the anchor and another specific for a sequence within the molecule of interest. The anchor oligonucleotide has been especially designed to facilitate subsequent analysis and cloning of the resultant PCR products. This three-stage procedure does not require purification of product between steps and avoids many of the technical difficulties associated with established anchored PCR protocols. The efficacy of ligation-anchored PCR was demonstrated by amplification of a specific IgG1 cDNA; total RNA equivalent to as few as 100 cells yielded the expected PCR product.
Long PCR,
Introduction
Long range PCR allows the amplification of PCR products, which are much larger than those achieved with conventional Taq polymerases. Up to 27 kb fragments are possible from good quality genomic DNA, although 10 - 20 kb fragments are routinely achievable, given the appropriate conditions. The method relies on a mixture of thermo stable DNA polymerases, usually Taq DNA polymerase for high processivity (i.e. 5’-3’ polymerase activity) and another DNA polymerase with 3’-5’ proofreading abilities (usually Pwo). This combination of features allows longer primer extension than can be achieved with Taq alone.
This method for detection of the FVIII gene intron 22 inversion (Liu et al, 1998) removes the requirement for Southern Blotting. Results can be obtained within 24 hours. Modifications from standard long range PCR protocols include the addition of DMSO and incorporation of deaza GTP to enable read through of a high GC content region upstream of the FVIII gene. The method relies on overlapping PCR to generate a constant band, which appears in all template DNA’s. This band acts as a control to show that the reaction has worked efficiently. The largest amplification product seen using this method is 12 kb, well within the range of the enzyme mix utilised.
Marathon RNA PCR.
cDNA Amplification Kits for Rapid Amplification of cDNA Ends—5' RACE & 3' RACE
Rapid Amplification of cDNA Ends (RACE) is a technique used to obtain the full-length sequence of an RNA transcript found within a cell. RACE can provide the sequence of an RNA transcript from a small known sequence within the transcript all the way to the 5' end (5' RACE-PCR) or 3' end (3' RACE-PCR) of the RNA
RACE-Ready Mouse cDNA
Marathon-Ready cDNAs—cDNAs made from high-quality Premium Poly A+ RNA and ligated to the Marathon Adaptor—are ready for 5'- and 3'-RACE PCR (1). Each Marathon-Ready cDNA is a premade, tissue-specific “pool” of double-stranded cDNA from which full-length genes can be amplified by using sets of gene-specific primers. They can also be used to study tissue-specific gene expression and to find polymorphic forms of mRNA or mRNA belonging to a multigene family.
Nested PCR,
Nested polymerase chain reaction is a modification of polymerase chain reaction intended to reduce the contamination in products due to the amplification of unexpected primer binding sites.
Polymerase chain reaction itself is the process used to amplify DNA samples, via a temperature-mediated DNA polymerase. The products can be used for sequencing or analysis, and this process is a key part of many genetics research laboratories, along with uses in DNA fingerprinting for forensics and other human genetic cases. Conventional PCR requires primers complementary to the termini of the target DNA. A commonly occurring problem is primers binding to incorrect regions of the DNA, giving unexpected products.
Nested polymerase chain reaction involves two sets of primers, used in two successive runs of polymerase chain reaction, the second set intended to amplify a secondary target within the first run product.
Processes
- The target DNA undergoes the first run of polymerase chain reaction with the first set of primers, shown in green. The selection of alternative and similar primer binding sites gives a selection of products, only one containing the intended sequence.
- The product from the first reaction undergoes a second run with the second set of primers, shown in red. It is very unlikely that any of the unwanted PCR products contain binding sites for both the new primers, ensuring the product from the second PCR has little contamination from unwanted products of primer dimers, hairpins, and alternative primer target sequences.

http://www.ivpresearch.org/
Non-competitive RT-PCR.(from Molecular techniques and Methods)- From-(IKI).
Noncompetitive RT-PCR
relies on the observation that prior to the onset of the plateau effect there
is a linear relationship between the quantity of input RNA and final product
during PCR amplification. To determine the number of cycles at which this
linear relationship occurs, the initial sample of RNA should express high
levels of target mRNA. Exogenous target should not be added to the RNA sample;
instead, the sample should have endogenous target gene expression that is high,
but no more than two to three times the highest levels that would be expected
in an actual experiment.
Using this procedure, one can obtain three levels of specificity: (1)
amplification of the product with specific primers; (2) correspondence of the
actual product size to the original estimated product size; and (3) hybridization
of the product with an internal probe not corresponding to either primer. In
addition, the low cycle number reduces artifacts such as nonspecific
amplifications that could pose major obstacles at higher cycle numbers. Another
technique for detecting the product involves the incorporation of labeled
nucleotides into PCR products that are then resolved by gel electrophoresis.
However, this approach is often associated with trace amounts of unincorporated
label that can produce a "trail" of label throughout the lane of an
electrophoretic gel. One can also use labeled primers at the beginning of the
assay or as a final PCR step with a new internal labeled primer annealed to one
strand of the PCR product and extended using Taq DNA polymerase, but both
approaches often result in considerable background radioactivity and rarely
give signals as well-defined and quantifiable as does the Southern blot.
· PCR cloning,
Since its discovery in 1983 the polymerase chain reaction has revolutionized molecular biology. Today, new forms of PCR and the related techniques of cloning are finding new applications at the cutting edge of biomedicine. by Peter Gwynne and Gary Heebner. PCR amplification of a segment of DNA produces 3’T ends. This DNA segment can be ligated to suitable vectors which have complementary ends.

PCR Cloning System
The Clontech In-Fusion® PCR Cloning System allows you to fuse of the ends of the PCR fragment to the homologous ends of a linearized vector. The 3' and 5' regions of homology are generated by adding 15 bp extensions to both PCR primers that precisely match the ends of the linearized vector. When the vector is combined with your insert, the In-Fusion® enzyme converts the double-stranded extensions into single-stranded DNA and fuses these regions to the corresponding ends of the linearized vector.
RACE-PCR.
Rapid Amplification of cDNA Ends (RACE) is a technique used in molecular biology to obtain the full length sequence of an RNA transcript found within a cell. RACE results in the production of a cDNA copy of the RNA sequence of interest, produced through reverse transcription, followed by PCR amplification of the cDNA copies (see RT-PCR). The amplified cDNA copies are then sequenced and, if long enough, should map to a unique mRNA already described, the full sequence of which is known. RACE can provide the sequence of an RNA transcript from a small known sequence within the transcript to the 5' end (5' RACE-PCR) or 3' end (3' RACE-PCR) of the RNA. This technique is sometimes called one-sided PCR or anchored PCR.
The first step in RACE is to use reverse transcription to produce a cDNA copy of a region of the RNA transcript. In this process, an unknown end portion of a transcript is copied using a known sequence from the center of the transcript. The copied region is bounded by the known sequence, and either the 5' or 3' end.
The protocols for 5' or 3' RACES differ slightly. 5' RACE-PCR begins using mRNA as a template for a first round of cDNA synthesis (or reverse transcription) reaction using an anti-sense (reverse) oligonucleotide primer that recognizes a known sequence in the gene of interest; the primer is called a gene specific primer (GSP), and it copies the mRNA template in the 3' to the 5' direction to generate a specific single-stranded cDNA product. Following cDNA synthesis, the enzyme terminal deoxy nucleotidyl transferase (TdT) is used to add a string of identical nucleotides, known as a homopolymeric tail, to the 3' end of the cDNA. (There are some other ways to add the 3'-terminal sequence for the first strand of the de novo cDNA synthesised which are much more efficient than homopolymeric tailing, but the sense of the method remains the same). A PCR reaction is then carried out, which uses a second anti-sense gene specific primer (GSP2) that binds to the known sequence, and a sense (forward) universal primer (UP) that binds the homopolymeric tail added to the 3' ends of the cDNAs to amplify a cDNA product from the 5' end.
3' RACE-PCR uses the natural polyA tail that exists at the 3' end of all eukaryotic mRNAs for priming during reverse transcription, so this method does not require the addition of nucleotides by TdT. cDNAs are generated using an Oligo-dT-adaptor primer that complements the polyA stretch and adds a special adaptor sequence to the 5' end of each cDNA. PCR is then used to amplify 3' cDNA from a known region using a sense GSP, and an anti-sense primer complementary to the adaptor sequence (wikipedia).
RAP-PCR (RNA arbitrarily primed PCR);
Screening of Differentially Amplified cDNA Products from RNA Arbitrarily Primed PCR Fingerprints Using Single Strand Conformation Polymorphism (SSCP) Gels ; Françoise Mathieu-Daudé, Rita Cheng+, John Welsh* and Michael McClelland.
Arbitrarily primed PCR fingerprinting of RNA and differential display resolved on an acrylamide gel has been extensively used to detect differentially expressed RNAs. However, after a differentially amplified product is detected the next steps are labor-intensive: a small portion of the fingerprinting gel that contains the differentially amplified product is cut out, reamplified and the correct product is determined, typically by cloning and sequencing what is often a mixture of products of similar size. Here we use a native acrylamide gel to separate DNAs in the reamplified mixture based on single-stranded conformation polymorphisms. Reamplifications are performed for the region carrying the differentially amplified product and a corresponding region from an adjacent lane where the product is less prominent or not visible. Denaturation of the reamplified DNA followed by side-by-side comparison on an SSCP gel allows the classification of reamplified material into (i) those that can be directly cloned because the differentially amplified product is relatively pure, (ii) those that need to be reamplified from the SSCP gel before cloning and (iii) those that are too complex for further study. This screen should save considerable effort now wasted on directly cloning unsuitable products from RNA fingerprinting experiments. An example is presented of cloning a gene differentially expressed in Trypanosoma brucei life cycle
RNase ligase mediated PCR (RLM_RACE PCR).
Mapping the 5′ and 3′ ends of Tetrahymena thermophila mRNAs using RNA ligase mediated amplification of cDNA ends (RLM-RACE), Xiuwen Liu and Martin A. Gorovsky*.
Procedure is described for mapping the ends of RNAs. Using T4 RNA ligase, a DNA (3′ end) or RNA (5′ end) oligonucleotide is ligated to RNA ends followed by cDNA synthesis, PCR amplification, cloning and sequencing. This method determines 5′ ends, 3′ polyadenylation sites and the size of poly(A) tails, and should be applicable to non-polyadenylated mRNAs and to non-message RNAs. Analysis of four Tetrahymena thermophila histone mRNAs revealed multiple closely spaced 5′ ends consistent with those determined by other methods. Except for a ′CCAAT′ box in either orientation 100–200 nucleotides upstream of the transcription start site, no conserved sequence elements were observed in the untranslated 5′ region or in sequences immediately flanking the transcription start site. Analysis of the 3′ ends of mRNAs encoding four histones, two tubutins and the Tetrahymena TATA binding protein confirmed the observations that Tetrahymena histone messages are polyadenylated and that poly(A) tails in this organism are short (ñ 50 nt). No canonical poly(A) addition signal was identified. The four histone messages analyzed here contained three sequence elements, TGTGT-TAAAAGTATT, not found in non-histone messages. Two non-histone messages contained GCATT(N)15ATACC near the poly(A) addition site.
· Multiplex-PCR uses several pairs of primers annealing to different target sequences. This permits the simultaneous analysis of multiple targets in a single sample. For example, in testing for genetic mutations, six or more amplifications might be combined. In the standard protocol for DNA Fingerprinting, the targets assayed are often amplified in groups of 3 or 4. Multiplex Ligation-dependent Probe Amplification (or MLPA) permits multiple targets to be amplified using only a single pair of primers, avoiding the resolution limitations of multiplex PCR. Multiplex PCR has also been used for analysis of microsatellites and SNPs.
Real-time polymerase chain reaction:
From Wikipedia, the free encyclopedia
(Redirected from Q-PCR), Jump to: navigation, search ,For reverse transcription polymerase chain reaction (RT-PCR), see reverse transcription polymerase chain reaction.
In molecular biology, real-time polymerase chain reaction, also called quantitative real time polymerase chain reaction (Q-PCR/qPCR/qrt-PCR) or kinetic polymerase chain reaction (KPCR), is a laboratory technique based on the PCR, which is used to amplify and simultaneously quantify a targeted DNA molecule. For one or more specific sequences in a DNA sample, Real Time-PCR enables both detection and quantification
.The quantity can be either an absolute number of copies or a relative amount when normalized to DNA input or additional normalizing genes.
The procedure follows the general principle of polymerase chain reaction; its key feature is that the amplified DNA is detected as the reaction progresses in real time. This is a new approach compared to standard PCR, where the product of the reaction is detected at its end. Two common methods for detection of products in real-time PCR are: (1) non-specific fluorescent dyes that intercalate with any double-stranded DNA, and (2) sequence-specific DNA probes consisting of oligonucleotides that are labeled with a fluorescent reporter which permits detection only after hybridization of the probe with its complementary DNA target.
Frequently, real-time PCR is combined with reverse transcription to quantify messenger RNA and Non-coding RNA in cells or tissues.
Abbreviations used for real-time PCR methods vary widely and include: RTQ-PCR, Q-PCR or qPCR.[1] Real-time reverse-transcription PCR is often denoted as: qRT-PCR,[2] RRT-PCR,[3] or RT-rt PCR.[4] The acronym "RT-PCR" commonly denotes reverse-transcription PCR and not real-time PCR, but not all authors adhere to this convention.[5]
RT PCR,
Reverse transcription polymerase chain reaction (RT-PCR) is a variant of polymerase chain reaction (PCR), a laboratory technique commonly used in molecular biology to generate many copies of a DNA sequence, a process termed "amplification". In RT-PCR, however, an RNA strand is first reverse transcribed into its DNA complement (complementary DNA, or cDNA) using the enzyme reverse transcriptase, and the resulting cDNA is amplified using traditional PCR or real-time PCR. Reverse transcription PCR is not to be confused with real-time polymerase chain reaction (Q-PCR/qRT-PCR), which is also sometimes abbreviated as RT-PCR.
For more details on this topic, see Reverse transcription polymerase chain reaction.
Reverse transcriptase is commonly used in research to apply the polymerase chain reaction technique to RNA in a technique called reverse transcription polymerase chain reaction (RT-PCR). The classical PCR technique can be applied only to DNA strands, but, with the help of reverse transcriptase, RNA can be transcribed into DNA, thus making PCR analysis of RNA molecules possible. Reverse transcriptase is used also to create cDNA libraries from mRNA. The commercial availability of reverse transcriptase greatly improved knowledge in the area of molecular biology, as, along with other enzymes, it allowed scientists to clone, sequence, and characterize DNA.
Reverse transcriptase has also been employed in insulin production. By inserting eukaryotic mRNA for insulin production along with reverse transcriptase into bacteria, the mRNA can insert itself into the prokaryote's genome, and large amounts of insulin can be created, sidestepping the need to harvest pig pancreas and other such traditional sources. Inserting eukaryotic DNA (instead of mRNA) into bacteria would not work because it is fragmented, with introns, and would not transcribe successfully using the bacteria's ribosomes.
Frequently, real-time PCR is combined with reverse transcription to quantify messenger RNA and Non-coding RNA in cells or tissues.
Abbreviations used for real-time PCR methods vary widely and include: RTQ-PCR, Q-PCR or qPCR. Real-time reverse-transcription PCR is often denoted as: qRT-PCR, RRT-PCR, or RT-rt PCR. The acronym "RT-PCR" commonly denotes reverse-transcription PCR and not real-time PCR, but not all authors adhere to this convention.
Quantitative PCR (or Q-PCR) is used to measure the specific amount of target DNA (or RNA) in a sample. By measuring amplification only within the phase of true exponential increase, the amount of measured product more accurately reflects the initial amount of target. Special thermal cyclers are used that monitor the amount of product during the amplification. Quantitative Real-Time PCR (QRT-PCR) methods use fluorescent dyes, such as Sybr Green, or fluorophore-containing DNA probes, such as TaqMan, to measure the amount of amplified product as the amplification progresses.
Nested PCR

Nested PCR:Asymmetric PCR:
Nested PCR increase the specificity of DNA amplification. Two sets of primers are used in two successive reactions. In the first PCR, one pair of primers is used to generate DNA products, which may contain products amplified from non-target areas. The products from the first PCR are then used as template in a second PCR, using one ('hemi-nesting') or two different primers whose binding sites are located (nested) within the first set, thus increasing specificity. Nested PCR is often more successful in specifically amplifying long DNA products than conventional PCR, but it requires more detailed knowledge of the sequence of the target.
Often only a small modification needs to be made to the standard PCR protocol to achieve a desired goal:
Multiplex-PCR uses several pairs of primers annealing to different target sequences. This permits the simultaneous analysis of multiple targets RT PCR,
a single sample. For example, in testing for genetic mutations, six or more amplifications might be combined. In the standard protocol for DNA Fingerprinting, the targets assayed are often amplified in groups of 3 or 4. Multiplex Ligation-dependent Probe Amplification (or MLPA) permits multiple targets to be amplified using only a single pair of primers, avoiding the resolution limitations of multiplex PCR. Multiplex PCR has also been used for analysis of microsatellites and SNPs.
VNTR PCR: Variations in VNTR lengths in 6 individuals.
· Variable Number of Tandem Repeats (VNTR) PCR targets areas of the genome that exhibit length variation. The analysis of the genotypes of the sample usually involves sizing of the amplification products by gel electrophoresis. Analysis of smaller VNTR segments known as Short Tandem Repeats (or STRs) is the basis for DNA Fingerprinting databases such as CODIS. http://en.wikipedia.org/w

VNTR PCR DNA fragments on Gel
Asymmetric PCR preferentially amplifies one strand of the target DNA. It is used in some sequencing methods and hybridization probing, to generate one DNA strand as product. Thermocycling is carried out as in PCR, but with a limiting amount or leaving out one of the primers. When the limiting primer becomes depleted, replication increases arithmetically through extension of the excess primer. A modification of this process, named L'inear-After-The-Exponential-PCR (or LATE-PCR), uses a limiting primer with a higher Melting temperature (Tm) than the excess primer to maintain reaction efficiency as the limiting primer concentration decreases mid-reaction. (Also see Overlap-extension PCR).
· Some modifications are needed to perform long PCR. The original Klenow-based PCR process did not generate products that were larger than about 400 bp. Taq polymerase can however amplify targets of up to several thousand bp long. Since then, modified protocols with Taq enzyme have allowed targets of over 50 kb to be amplified.[5]
· Quantitative PCR (or Q-PCR) is used to measure the specific amount of target DNA (or RNA) in a sample. By measuring amplification only within the phase of true exponential increase, the amount of measured product more accurately reflects the initial amount of target. Special thermal cyclers are used that monitor the amount of product during the amplification. Quantitative Real-Time PCR (QRT-PCR) methods use fluorescent dyes, such as Sybr Green, or fluorophore-containing DNA probes, such as TaqMan, to measure the amount of amplified product as the amplification progresses.
· Hot-start PCR is a technique performed manually by heating the reaction components to the DNA melting temperature (e.g. 95°C) before adding the polymerase. In this way, non-specific amplification at lower temperatures is prevented. Alternatively, specialized reagents inhibit the polymerase's activity at ambient temperature, either by the binding of an antibody, or by the presence of covalently bound inhibitors that only dissociate after a high-temperature activation step. 'Hot-start/cold-finish PCR' is achieved with new hybrid polymerases that are inactive at ambient temperature and are only activated at elevated temperatures.
· In Touchdown PCR, the annealing temperature is gradually decreased in later cycles. The annealing temperature in the early cycles is usually 3-5°C above the standard Tm of the primers used, while in the later cycles it is a similar amount below the Tm. The initial higher annealing temperature leads to greater specificity for primer binding, while the lower temperatures permit more efficient amplification at the end of the reaction.
· Assembly PCR (also known as Polymerase Cycling Assembly or PCA) is the synthesis of long DNA structures by performing PCR on a pool of long oligonucleotides with short overlapping segments, to assemble two or more pieces of DNA into one piece. It involves an initial PCR with primers that have an overlap and a second PCR using the products as the template that generates the final full-length product. This technique may substitute for ligation-based assembly.
· In Colony PCR, bacterial colonies are screened directly by PCR, for example, the screen for correct DNA vector constructs. Colonies are sampled with a sterile pipette tip and a small quantity of cells transferred into a PCR mix. To release the DNA from the cells, the PCR is either started with an extended time at 95°C (when standard polymerase is used), or with a shortened denaturation step at 100°C and special chimeric DNA polymerase.[9]
· The Digital polymerase chain reaction simultaneously amplifies thousands of samples, each in a separate droplet within an emulsion.
Pretreatments and extensions:
The basic PCR process can sometimes precede or follow another technique:
· RT-PCR (or Reverse Transcription PCR) is used to reverse-transcribe and amplify RNA to cDNA. PCR is preceded by a reaction using reverse transcriptase, an enzyme that converts RNA into cDNA. The two reactions may be combined in a tube, with the initial heating step of PCR being used to inactivate the transcriptase.[4] The Tth polymerase (described below) has RT activity, and can carry out the entire reaction. RT-PCR is widely used in expression profiling, which detects the expression of a gene. It can also be used to obtain sequence of an RNA transcript, which may aid the determination of the transcription start and termination sites (by RACE-PCR) and facilitate mapping of the location of exons and introns in a gene sequence.
· Ligation-mediated PCR uses small DNA oligonucleotide 'linkers' (or adaptors) that are first ligated to fragments of the target DNA. PCR primers that anneal to the linker sequences are then used to amplify the target fragments. This method is deployed for DNA sequencing, genome walking, and DNA footprinting.[10] A related technique is Amplified fragment length polymorphism, which generates diagnostic fragments of a genome.
· Methylation-specific PCR (MSP) is used to identify patterns of DNA methylation at cytosine-guanine (CpG) islands in genomic DNA.[11] Target DNA is first treated with sodium bisulfite, which converts unmethylated cytosine bases to uracil, which is complementary to adenosine in PCR primers. Two amplifications are then carried out on the bisulfite-treated DNA: One primer set anneals to DNA with cytosines (corresponding to methylated cytosine), and the other set anneals to DNA with uracil (corresponding to unmethylated cytosine). MSP used in Q-PCR provides quantitative information about the methylation state of a given CpG island.

Mol Biol
Application of PCR:
This technique has been widely used for variety of purposes and so it is considered as the most versatile methodology used in all most all areas of biosciences and medicine.
· Screening of cDNA library using a specific primer.
· One can amplify a specific species of mRNA out of thousands on mRNAs by using species-specific primers.
· It can be used to amplify only one strand for using it as a probe.
· With designed primers with restriction sites it is possible to amplify a DNA segment and clone in such a way it can produce either RNA or anti sense RNAs.
· It can be used for site directed mutagenesis, which is used for protein engineering techniques.
· PCR can be used for ssDNA or dsDNA sequencing, in fact in most of the labs the PCR sequencing is routine. A substantial molecular evolutionists use PCR based sequencing for tracing inter relationship among the members and molecular evolutionary trends.
· Used for detecting infectious agents or pathogen provided the sequence of each of the pathogens are known. It is very easy to diagnose malarial parasite infection, other wise it takes six to eight days.
· It can also be used detecting genetic diseases by employing know primers for specific group of Exons.
· HIV disease can be identified to 99% accuracy and certainty. It is quicker and cost effective.
· Used for sex determination at neonatal stage by using floating embryonic cells. The Y-chromosomes carries 3.5 Kbp long DYZ-1 sequence in 5000 copies. A consensus sequence of 149 bp long can be detected by PCR.
· This technique is also used to detect X –linked inherited disorders.
· One can amplify DNA from mummies and fossils.
· PCR can be employed to detect RFLP.
· Used for identifying linkage mapping.
· It is extensively used in RAPD analysis for detecting difference among the members of a population. In fact it is used detect quantitative traits.
· This can also be used for single strand nucleotide polymorphism.
· PCR amplified DNA can directly used for cloning into a proper vectors. This is because; when the DNA is amplified one finds one nucleotide ‘A’ at 3’ end as an overhang. Such DNAs can be cloned into vectors containing ‘T’ as overhangs.
In recent times PCR is used for DNA profiling or what is called genetic finger printing, which is useful in solving parental dispute and also greatly helps in forensic detection. The same technology can extended to identify the relationship of the lost or missing persons.

Genetic finger profiling- a PCR machine used

DNA profiling
RACE PCR:

RT PCR

The reverse transcriptase-polymerase chain reaction (RT-PCR) technique for determining whether a particular type of mRNA is present. First, the mRNA from a small sample is converted to double-stranded cDNA using the enzymes reverse transcriptase and RNase H. A primer is added to the cDNA and the second strand is completed using thermostable DNA polymerase from T. aquaticus (Taq polymerase). This “target” DNA is then denatured and two sets of primers are added; the primers hybridize to opposite ends of the target sequence if the sequence is present. (This will happen only if the mRNA of interest was originally present.) When Taq polymerase is added to the denatured DNA, each strand synthesizes its complement. http://10e.devbio.com/


RT PCR
PCR cloning:
T-A Cloning Strategy: Taq and other polymerases seem to have a terminal transferase activity which results in the non-templated addition of a single nucleotide to the 3'-ends of PCR products. In the presence of all 4 dNTPs, dA is preferentially added; however, use of a single dNTP in a reaction mix results in (relatively inefficient) addition of that nucleotide. This complicates cloning, as the supposedly blunt-ended PCR product often is not, and blunt-ended cloning protocols often do not work or are very inefficient. This can be remedied by incubation of PCR products with T4 DNA pol or Klenow pol, which "polishes" the ends due to a 3'->5' exonuclease activity (Lui and Schwartz, 1992; BioTechniques, 20: 28-30). However, this terminal transferase activity is also the basis of a clever cloning strategy: this uses Taq pol to add a single dT to the 3'-ends of a blunt-cut cloning vector such as pUC or pBluescriptTM, and simple ligation of the PCR product into the now "sticky-ended" plasmid (Marchuk et al., 1990; NAR 19: 1156).

PCR-TA cloning or PCR cloning

PCR cloning protocol
Real time PCR:
Real-time PCR is able to detect sequence-specific PCR products as they accumulate in "real-time" during the PCR amplification process. As the PCR product of interest is produced, real-time PCR can detect their accumulation and quantify the number of substrates present in the initial PCR mixture before amplification began.
There are a few different variations of the procedure, but the one illustrated here is called molecular beacon<www.molecular-beacons.org/>. Molecular beacons are short segments of single-stranded DNA (Figure 1). The sequence of each molecular beacon must be customized to detect the PCR product of interest. In figure one, you can see there are nine bases on one end of the molecular beacon that can base pair with nine bases on the other end of the beacon. This complementation permits the molecular beacon to form a hairpin structure. The loop portion of the molecular beacon is composed of bases (shown as pink lines) that are complementary to one strand of the PCR product the investigator wants to detect and quantify. http://www.bio.davidson.edu/
Attached to opposite ends of the beacon are a fluorescent reporter dye and a quencher dye. When the molecular beacon is in the hairpin conformation, any fluorescence emitted by the reporter is absorbed by the quencher dye and no fluorescence is detected.

Diagram of molecular beacon. This beacon is 33 nucleotides long with a reporter dye attached to the 5' end and a quencher attached to the 3' end. The nine 5' bases are able to form base pairs with the nine 3' bases which brings the reporter and quencher in very close proximity. Therefore, when the reporter is excited by the appropriate light, its emission is absorbed by the quencher and no fluorescence is detected. The pink lines represent nucleotides that can form base pairs with the PCR product under investigation. http://www.bio.davidson.edu/
The PCR portion of real-time PCR is standard. Two PCR primers are used to amplify a segment of DNA (Figure 2).

PCR product of interest. The two primers are show as purple arrows and the base pairing between the two strands are shown in pink. http://www.bio.davidson.edu/

Detection of PCR product by molecular beacon. When the beacon binds to the PCR product, it is able to fluoresce when excited by the appropriate wavelength of light. The amount of fluorescence is directly proportional to the amount of PCR product amplified; http://www.bio.davidson.edu/
ReL TIME PCR; The Taq Man method;

The Taqman probe. The red circle represents the quenching dye that disrupts the observable signal from the reporter dye (green circle) when it is within a short distance. Image created by Dan Pierce. http://www.bio.davidson.edu/
The probe consists of two types of fluorophores, which are the fluorescent parts of reporter proteins (Green Fluorescent Protein (GFP) has an often-used fluorophore). While the probe is attached or unattached to the template DNA and before the polymerase acts, the quencher (Q) fluorophore (usually a long-wavelength colored dye, such as red) reduces the fluorescence from the reporter (R) fluorophore (usually a short-wavelength colored dye, such as green). It does this by the use of Fluorescence (or Förster) Resonance Energy Transfer (FRET), which is the inhibition of one dye caused by another without emission of a proton. The reporter dye is found on the 5’ end of the probe and the quencher at the 3’ end. (www.probes.com2003); http://www.bio.davidson.edu/

The TaqMan® probe binds to the target DNA, and the primer binds as well. Because the primer is bound, Taqpolymerase can now create a complementary strand. Image created by Dan Pierce. http://www.bio.davidson.edu/
Once the TaqMan® probe has bound to its specific piece of the template DNA after denaturation (high temperature) and the reaction cools, the primers anneal to the DNA. Taq polymerase then adds nucleotides and removes the Taqman® probe from the template DNA. This separates the quencher from the reporter, and allows the reporter to give off its emit its energy. This is then quantified using a computer. The more times the denaturing and annealing takes place, the more opportunities there are for the Taqman® probe to bind and, in turn, the more emitted light is detected. (www.probes.com 2003); http://www.bio.davidson.edu/
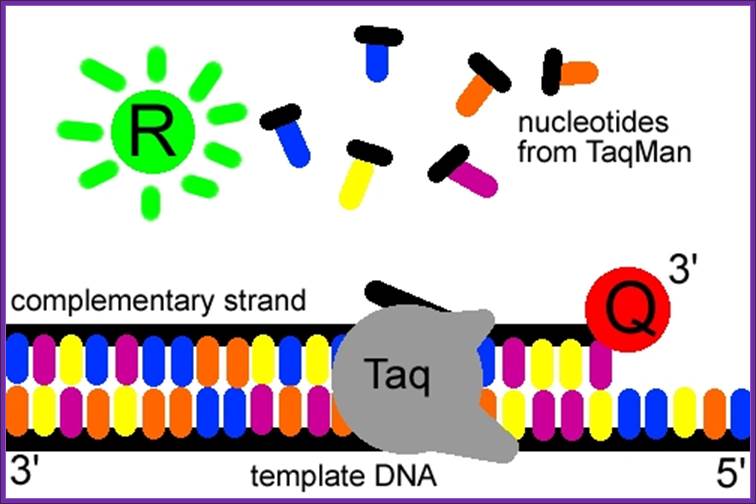
The reporter dye is released from the extending double-stranded DNA created by the Taq polymerase. Away from the quenching dye, the light emitted from the reporter dye in an excited state can now be observed. Image created by Dan Pierce. http://www.bio.davidson.edu/
Multiplex PCR;

Multiplex PCR

Multiplex PCR for detection of plasmid-mediated quinolone resistance qnr genes in ESBL-producing enterobacterial isolates; http://jac.oxfordjournals.org/

(a) The first-step multiplex PCR requires two sets of primers, A and B, that are used in independent multiplex PCRs. Together, primer sets A and B cover the sequence of interest. Within each set PCR fragments do not overlap. However, PCR fragments between set A and B show overlap of non-primer-determined sequences. (b–d) Second-step simplex PCRs are used for individual reamplification of each of the fragments obtained in the multiplex PCR. The strategy in b uses the same primer pairs used in the multiplex sets. In the strategy shown in c, two PCRs with nested primers on one end are used. Both strategies are suitable for amplification of fragmented DNA (>80 bp). In the strategy shown in d, completely nested primer pairs are used in the simplex PCRs. This two-sided nested variation can only be used for less fragmented DNA (>160 bp).Multiplex PCR; Holger Römpler et al; http://www.nature.com/

Multiplex PCR
.

With the current generation of multiplex rt-PCR cyclers that can employ five simultaneous fluorescence colours (four "experimental" plus a control), the four tests can be run simultaneously, with a four-fold gain in throughput.
https://www.mun.ca
PCR based genetic analysis

Site specific in vitro mutation by PCR

Cancer detection by PCR