Molecular Techniques:
DNA Synthesis and Sequencing:
DNA Synthesis:
Synthesis of DNA in vitro has its origin in the labs of Alexander Todd. The work has started as an in vitro synthesis of nucleosides and nucleotides. This was continued in many labs, but in the lab of H.G Khorana, it is culminated in the synthesis of tRNA gene, the first gene to be synthesized in the lab. Today the technology has so improved any DNA of any sequence can be automated for the synthesis. A technician without the knowledge of the chemical basis of synthesis can do this kind of work with some sophisticated machines.
To begin with the bases of dATP, dGTP and dCTP are derivatised by a process called Benzoylation, and there is no need to derivatise dTTP for it has no amino group. The 5’ CH2OH group of each nucleotide is added with Dimethoxytrityl (DMT). The 3’ phosphate group of the ribose is added with Diisopropylamine or 2-cyanomethyl group and a methyl group to the free end of the phosphate. Derivatization of the bases, DM- tritilation of the 5’ end and addition of Di isopropyl amine at the 3’ phosphate can be used for synthesis of oligo's.

Synthesis by phoshoramidite phosphonamidite
method; http://library.med.utah.edu/;https://en.wikipedia.org[KG1] [KG2] [KG3]
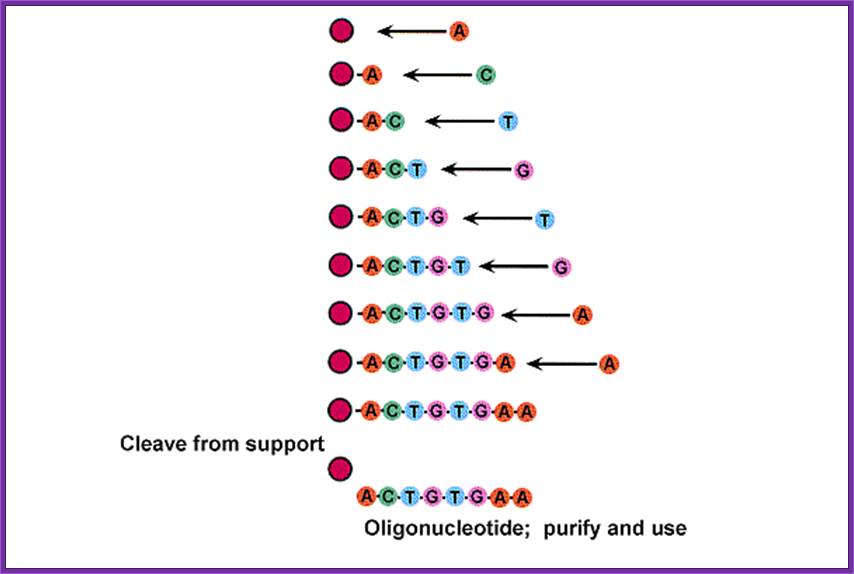
· To start with the first nucleotide at its 3’ OH of the sugar is linked to CPG (i.e. Controlled Pore Glass) with a spacer and the start nucleoside is benzoylated at the base and DMT-titillated at 5’ end. This nucleotide acts as the first nucleotide at the 3’ end of the oligo.
· Such nucleosides bound glass beads are loaded on to a special column and the quantity loaded should be noted.
· The column should be washed with Acetonitril to remove any other nucleophiles and water if any. This column is flushed with argon a noble gas or inert gas to remove Acetonitril.
· The column is treated with Trichloroacetic acid (TCA) to remove DMT and make the 5 CH2OH group free for activity. Then the column is washed with Acetonitril to remove any traces of TCA. Then it s flushed with Argon.
Now the 5’ end of the first nucleoside is ready for the addition of the second nucleotide to generate 5’à3’ phosphodiester link.
· A defined derivatised (DMT-tritylated and Di-isopropylated) nucleosides are added along with Tetrazole. The Tetrazole activates the coupling by removing the Diisopropylamine group from the 3’ end of the added nucleoside and facilitates the linking it to the first nucleoside by forming 2HC5’-O-P-O-3’ribose of the second nucleoside, where the methylated phoshphite is generated in between
Protected
2’deoxynucleoside phophoramdites; ;https://en.wikipedia.org/wiki
the first and the second nucleoside coupling.
· As the phosphate is very unstable and reactive, the column is added with Iodine to oxidize the phosphate to generate pentavalent phosphate group.
If the concentration of the second phosphoramidite added is in proper ratio and the reaction is properly controlled, it should achieve 98 to 98% of coupling.


· The column is washed with acetonitril and flushed with Argon.
· In this reaction not all the first nucleosides are linked to the second nucleoside, so they are rendered useless by blocking their 5’ end by adding acetic anhydride and dimethylaminopyridine. This reaction adds acetate group to the free 5’ group. This process is called capping and it prevents any addition of nucleotides in the next reaction.
· After this step the column is washed with Acetonitril and flushed with Argon.

This diagram is a generalized cycle,

Cycle of nucleosides addition

steps
This cycle of processes is repeated and can be done with automation. And the nucleotide sequences to be added are programmed. And quantity of it to be added and other washing and reactions and the reaction time can be programmed, so the entire synthesis of 50 or more nucleotide long oligo’s can be synthesized in a couple of hours.

Simplistic representation dinucleotide synthesis

Oligos are synthesized on digital plates for micro array protocols

DNA and RNA synthesizer; http://www.ucalgary.ca/
DNA synthesizer
· The methyl groups at the Phosphate, Benzoyl group at the base are removed by another set of chemical reactions.
· The glass beads at the first nucleotide at 3’end and DMTr at the 5’ end of the last nucleotide are removed by incubating the Oligo’s glass tube tightly capped in 50% Liquid ammonia at 55^oC overnight in a fume hood overnight.
· Cool the tube and spin at 10K for 5-8 minutes and the supernatant is carefully decanted and the pellet is lyophilized. The dried Oligo’s pellet is suspended in a desired buffer and purified on spin columns to get uniform sized Oligo’s.
One OD of oligo’s at 260nm gives a quantity of 30-33 ug per ml.
One OD of oligo’s at 260nm gives a quantity of 30-33 ug per ml.
The 5’ ends of the oligo’s don’t contain phosphate groups, if required they have to be phosphorylated at their 5’ end by using 5’Polynucleotide kinase reactions.

DNA/RNA/LNA Synthesizer H-32
Application:
Used in synthesizing a gene.
Used in preparing linkers and primer and primer adopters.
Used for the preparation of primers for various purposes.
Used for the preparation probes.
Used to create site directed mutagenesis.
Used to produce promoters.
Used to produce transcriptional terminators.
Used for the synthesis of desired MCSs
Synthetic primers are extensively used in PCR.
Brand New! The Dr. Oligo 06 and 192 Synthesizer is fully automated one and two plate synthesizers for high throughput, automated oligonucleotide synthesizer:

Oligo synthesizer

Peptide synthesizer
DNA sequencing:
Preparation of ssDNA:
· For sequencing one can use ssDNA or dsDNA.
· Use a bacterial strain that contains phagmid having the DNA, which is to be sequenced.
· Pick a colony and place it into 500ml LB flask containing Ampicillin. Grow cells overnight.
· Then transfer 2.5ml of cells into a flask containing 30 ml LB broth and Ampicillin.
· Inoculate the flask with previously prepared M13K07 helper phage.
· Shake the culture at 37^oC for 1 hr. Then add 100ug/ml Kanamycin and grow over night.
· Pellet the cells. Take the supernatant and add 30% PEG (PEG dissolved in 2.5M NaCl) to 8%, and allow to precipitate.
· Spin it for 5minutes at 12K and suspend the pellet in 1ml of TE.
· Extract the suspension with phenol: chloroform method. Take aqueous layer out and then precipitate the DNA with 2x ethanol or Isopropanol. Use Ammonium acetate for precipitation.
· The DNA recovered from this procedure is all single stranded.
Sanger’s chain elongation /termination protocol:
Denaturing reaction:
· 4-5ug of DNA in TE is added to a solution containing 0.2M NaOH, 0.2mM EDTA,
· Add water to 50ul.
· Incubate at 37°C for 30 minutes.
· Then add 3M Na acetate 0.1Volume (5ul), and add ethanol 2-4 volume.
· Precipitate at -70 for 30minutes,
· Centrifuge at 15K for 15 minutes.
· Suspend the pellet in 70% ethanol, then spin and air dry the pellet.
· Suspend the denatured pellet in 7ul of H2O
· Add 2ul of 5x buffer,
· And add 1ul of primer to the total volume of 10ul.
· Heat it to 65-70°C for 10 minutes and slowly cool the solution for primers to anneal to their respective sequence.
5x annealing buffer:
200mM Tris-Cl 7.5,
100mM MgCl2,
250mM NaCl.
Stock solutions:
1M NaOH,
0.5M EDTA, pH8.0.
1M-sodium acetate, pH5.5
Stop mix:
95% fomamide20mM EDTA pH 8.0,
0.05% Bromophenol Blue,
0.05% Xylene Cyanol.
Heat the reaction mix at 95^oC for 3-4 minute and immediately cool and load 5ul of each into labeled wells in the sequencing gel.
Stock solutions:
40% Acrylamide solution:
380gm Acrylamide,
20gm Bis acrylamide (N' methylene bis-acrylamide),
Add water to a liter.
10x TBE pH 8.8 (1 liter):
540gm Tris-,
275gm Boric acid (absolutely pure0,
46.5gm Na-EDTA,
Add dd H2O (double distilled) to a liter.
10% Ammonium per sulphate,
TEMED stock,
Siliconising solution (Replicote).
Preparation of 5% Gel: This is where science and art in confluence. All the chemicals used should be of pure quality.
10ml TBE buffer (10x),
12.5ml Polyacrylamide (40% stock),
42gm Urea (pure),
99ml ddWater,
1ml 10% NH4) 6 SO4,
20ul TEMED.
Prepare dNTPs stock:
7.5uM each of dGTP, dCTP and dTTP.
Ready with 35SdATP 250uci/mMol.
DNA plus primer 10ul,
dNTPs mix 2ul
0.1MDTT 1.0ul,
35S dATP 1ul,
T7 DNA pol 1ul (1unit),
Total-with buffer-15ul.
Keep the mix on ice.
Elongation and termination reaction:
Prepare the termination mix and keep it ready. It consists of 80uM each of dNTPs + 50mM NaCl,
Label tubes as G, A, T and C.
To each of the tubes add 2.5ul of the termination mix.
Then add 8uM each of ddGTP, ddATP, ddTTP and ddCTP to their respective tubes. The dd means dideoxy nucleotides, which don’t contain 2’ and 3’ OH groups.
Take out the tube containing primed DNA with T7 DNA-pol and incubate at 37^0C for 5minutes.
Then immediately dispense 3.5ul of Enzyme reaction mix to each of the labeled tubes and incubate for another 3-5 minutes at room temperature.
Then add 5ul of stop mix to each tube.
Stop mix:
95% fomamide20mM EDTA pH 8.0,
0.05% Bromophenol Blue,
0.05% Xylene Cyanol.
Heat the reaction mix at 95^oC for 3-4 minute and immediately cool and load 5ul of each into labeled wells in the sequencing gel.
Gel Preparation:
This is where science and art in confluence. All the chemicals used should be of pure quality.
Stock solutions:
40% Acrylamide solution:
380gm Acrylamide,
20gm Bis acrylamide (N' methylene bis-acrylamide),
Add water to a liter.
10x TBE pH 8.8 (1 liter):
540gm Tris-,
275gm Boric acid (absolutely pure0,
46.5gm Na-EDTA,
Add dd H2O (double distilled) to a liter.
10% Ammonium per sulphate,
TEMED stock,
Siliconising solution (Replicote).
Mix well and degas and slowly add the solution taking care not to develop any bubbles in the gel anywhere. Immediately place the shark comb.
After polymerization switch on the power pack and allow the current to flow for at least 30 minutes at 100watts.
Then wash the wells with running buffer with the help of Pasteur’s pipette. Then load the wells with the DNA samples (5ul each).
The Bromophenol blue front indicates the size of 35bp and Xylene cyanol represents 130bp.
Switch the power and adjust the power input at 100 watts. When the Xylene cyanol front runs down, load another four samples to the next four slots. At the same time add ammonium acetate to 0.3 M concentration to the bottom buffer holder and run the gel till the BP-blue front reaches the end of the gel. Stop the power.
Carefully remove the top plate and place a dry x-ray (used) on the gel and carefully remove the x-ray film for the gel is now stuck to the X-ray film.

DNA sequencing System, for Gel electrophoresis

Then fix the gel in 10% acetic acid + 10% methanol for 20-30 minutes. This treatment removes most of the urea. Then dry the gel in Gel-dryer and place the dried gel in a cassette and label or mark the positions. Place a new unexposed x-ray film. Tightly close the Cassette and wrap it with Aluminum foil and place it in a deep freezer or at -70^oC. Then remove the film and develop the film in dark. Dry the film and read the sequence with a sequence reader.

TGCA
Read the sequence from the right hand bottom to the top, start from the right four lanes; use the sample and read the sequence from the bottom to the top- AGGCAGGGT ---
Theory part of the experiment:
One can use ssDNA or ds DNA for sequencing. Basically there are two methods, which have been traditionally employed for sequencing. But with the advanced technology automated sequencing protocols are used. The latest is Nov11th 2011 nanopore “Ion Torrent Personal Genome Machine (PGM)”.
Among the traditional methods the one that is first used is Maxam and Gilbert method, which is also called chemical method. The second method is called primer extension method or Sanger’s method.
In Sanger’s method the principle applied is simple. Once the DNA has
been identified, one has to have primers. Most of the pUC plasmids on either side of the cloned DNA contain MCS of known sequences, which can be used for developing primers. If the starting material is double stranded, then it has to be denatured to ss state by treating with 0.5M NaOH and then the solution is neutralized and DNA is precipitated. Thus obtained ssDNA or ssDNA obtained from phagmid are annealed with forward primers. Then the primers are extended using T7 DNA-pol for about 4-5 minutes and the extension stopped by adding respective dideoxy nucleotides to the labeled tubes like G, A, T, C. As dd Nucleotides doesn’t have 3’OH groups the enzyme cannot extend the synthesis, so the extension is stopped. In this reaction not all DNA molecules that added to the reaction tube start extending simultaneously. So a mixture of extended strands whose size range from one to 500 or more nucleotides, which are labeled with radioactive isotopes exist in the each of the tubes where the DNA synthesis is terminated at the said dNTPs. When each of the samples from each of the tubes is run on a denaturing polyacrylamide gel and drying the gel and it is autographed. Then one can observe a ladder of bands in the X-ray film in each of the slots. The extended strands with smallest number of nucleotides are found at the lower end and those with 500 or more are found at the top region of the gel. Thus one can read the sequence from the base to the top in a sequence
From the smallest to the longest strand synthesized. By identifying the sequence where the extension is terminated one can get the sequence. If use the primer for the bottom strand we get the sequence for top strand which is referred to as the + strand.
Let us take an example:
5’CTAGCTAGCTAGCTAG3’ who’s other strand sequence is 3’GATCGATCGATCGATC5’. One can develop primers for the lower stand. And if the primer is extended then the sequence one obtains is of the top strand from 5’ to 3’ direction. Read the sequence from the bottom of the gel towards the top of the gel
Dd*CTP
3’GATCGATCGATCGATC5’
--------5’C*
1. 5’CTAGC*
5’CTAGCTAGC*
5’CTAGCTAGCTAGC*

dd*TTP
3’GATCGATCGATCGATC
--------5’CT*
5’CTAGCT*
5’CTAGCTAGCT*
5’CTAGCTAGCTAGCT*

dd*ATP
3’GATCGATCGATCGATC
--------5’CTA*
5’CTAGCTA*
5’CTAGCTAGCTA*
5’CTAGCTAGCTAGCTA*

dd*GTP
3’GATCGATCGATCGATC
--------5’CTAG*
5’CTAGCTAG*
5’CTAGCTAGCTAG*
5’CTAGCTAGCTAGCTAG*

|
|
G |
A |
T |
C |
|
|
16 |
G |
|
|
|
G |
|
15 |
|
A |
|
|
A |
|
14 |
|
|
T |
|
T |
|
13 |
|
|
|
C |
C |
|
12 |
G |
|
|
|
G3’ |
|
11 |
|
A |
|
|
A |
|
10 |
|
|
T |
|
T |
|
9 |
|
|
|
C |
C |
|
8 |
G |
|
|
|
G |
|
7 |
|
A |
|
|
A |
|
6 |
|
|
T |
|
T |
|
5 |
|
|
|
C |
C |
|
4 |
G |
|
|
|
G |
|
3 |
|
A |
|
|
A |
|
2 |
|
|
T |
|
T |
|
1 |
|
|
|
C |
C5’ |
The final reading is 5’CTAGCTAGCTAGCTAG3’
The number of nucleotides that one can read can be any where 400 to 800, if all goes well with your protocol.
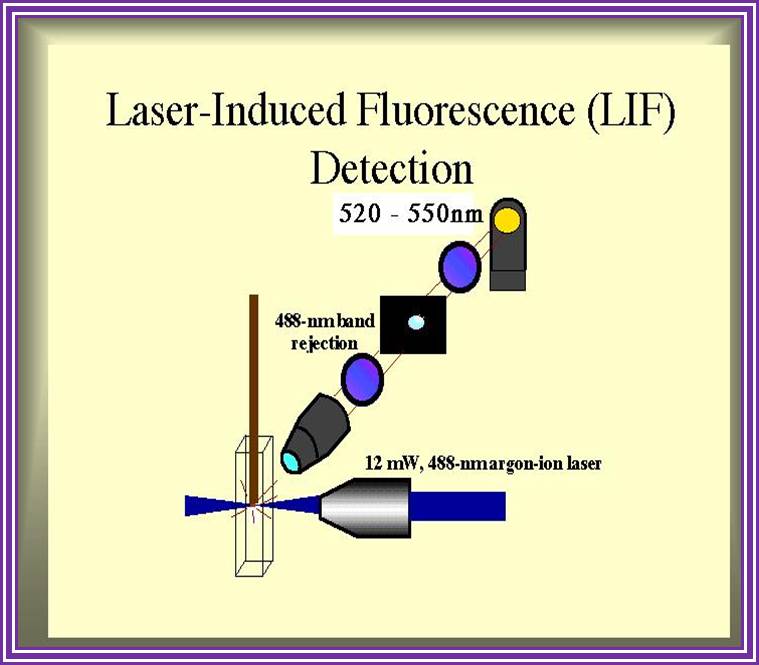
Now days the sequencing is automated and entire sequence is read from a single gel. One basic protocol is to use primers conjugated with different flourogeneic tags and one can load the all the reaction into a single lane and run the gel and read the sequence by the kind of flourogeneic color that each band emits on exposure to laser beams.
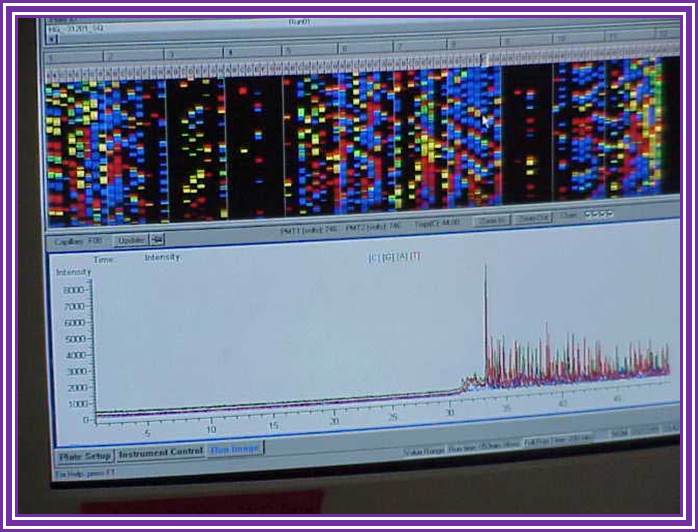
Now people use PCR protocols, where the entire reaction is initiated in a single tube and 30 to 32 rounds of amplification and termination is done. Loading the reaction samples into a single well will provide the sequence and the whole thing is so automated once you load the gel, one can go home and come back in the morning and get the sheet with the sequence typed. In recent years capillary electrophoresis method has been employed in DNA sequencing and it was found to be very efficient faster and accurate.
Routinely people sequence DNA using fluorescent labeled dideoxy nucleotides or fluorescent labeled primers. The reaction products are run on automated machines, where one can see the colored fragment moving down and the same are read by the computer and it takes out a print out at the end.
For fluorescent labeled sequencing dd ntds or primers are used. They are read by scanners and the data is recorded in computer hard disc
"G" tube: all four dNTP's, ddGTP and DNA polymerase
"A" tube: all four dNTP's, ddATP and DNA polymerase
"T" tube: all four dNTP's, ddTTP and DNA polymerase
"C" tube: all four dNTP's, ddCTP and DNA polymerase



http://www.bio.davidson.edu/

Sequences

An example of the results of automated chain-termination DNA sequencing; http://en.wikipedia.org/

Putting all four deoxynucleotides into the picture:
Well, OK, it's not so easy reading just C's, as you
perhaps saw in the last figure. The spacing between the bands isn't all that
easy to figure out. Imagine, though, that we ran the reaction with *all four*
of the dideoxy nucleotides (A, G, C and T) present, and with *different*
fluorescent colors on each. NOW look at the gel we'd get (at left). The
sequence of the DNA is rather obvious if you know the color codes ... just read
the colors from bottom to top: TGCGTCCA-(etc).http://seqcore.brcf.med.umich.edu/

RNA sequences




On a
large scale sequencing used for genomic studies capillary electrophoretic
system with the facility to run more than 390 reactions at any given time and
the time required is just under 3hrs and analyze 500 or more ntds. In a
singlea single day one can run six times,
each time let us say 300 columns, so 300 capillaries x 6 times x 500 bases, i.e
equivalent to sequencing 900,000 bases per day. The results are also
automatically read by computers and one gets the data sheet at every 2hrs or
so. This is an incredible technique, used in many labs who are engaged in
sequencing the genomes on large scale.
Sequencing using capillary electrophoresis:

Automated DNA sequencing by capillary electrophoresis; https://www.mun.ca


The process relies on nanopores, which are the go-to tech for companies trying to pull this off. Basically, a DNA strand is pushed through a 2nm hole on a silicon chip and, as it moves through, that chip is able to use an electrical charge to read the strand's coding sequence. That is then spit out to a supercomputer to crunch the numbers at a speed of 10 million bases per second and, within minutes, you too can have some hard data to make you freak out about the future. Nanopore DNA sequencing technique promises complete genome in minutes or your income behind: Posted in December 24th, 2010; Nanopore DNA sequencing technique promises entire genome in minutes or your money back; Tim Stevens, http://www.engadget.com/
Dye-terminator sequencing
An alternative to primer labeling the chain terminators are labeled, a method commonly called ‘dye-terminator sequencing’. The major advantage of this method is that the sequencing can be performed in a single reaction, rather than four reactions as in the labeled-primer method. In dye-terminator sequencing, each of the four dideoxynucleotide chain terminators is labeled with a different fluorescent dye, each fluorescing at a different wavelength. This method is attractive because of its greater expediency and speed and is now the mainstay in automated sequencing with computer-controlled sequence analyzers (see below). Its potential limitations include dye effects due to differences in the incorporation of the dye-labeled chain terminators into the DNA fragment, resulting in unequal peak heights and shapes in the electronic DNA sequence trace chromatogram after capillary electrophoresis (see figure to the right). This problem has largely been overcome with the introduction of new DNA polymerase enzyme systems and dyes that minimize incorporation variability, as well as methods for eliminating “dye blobs”, caused by certain chemical characteristics of the dyes that can result in artifacts in DNA sequence traces. The dye-terminator sequencing method, along with automated high-throughput DNA sequence analyzers, is now being used for the vast majority of sequencing projects, as it is both easier to perform and lower in cost than most previous sequencing methods.

Automation and sample preparation;
Modern automated DNA sequencing instruments (DNA sequencers) can sequence up to 384 fluorescently labeled samples in a single batch (run) and perform as many as 24 runs a day. However, automated DNA sequencers carry out only DNA size separation by capillary electrophoresis, detection and recording of dye fluorescence, and data output as fluorescent peak trace chromatograms. Sequencing reactions by thermo cycling, cleanup and re-suspension in a buffer solution before loading onto the sequencer are performed separately.

Figure: Sequence ladder by radioactive sequencing compared to fluorescent peaks

Figure: View of the start of an example dye-terminator read








Single-molecule real-time sequencing (SMRT®):

Called single-molecule real-time sequencing (SMRT®);
The technology promises genetic testing at under $200 per person, according to Bloomberg.
PacBio's SMRT technology is built upon two key innovations that overcome major challenges facing the field of DNA sequencing: The SMRT chip, which enables observation of individual fluorophore against a dense background of labeled nucleotides by maintaining a high signal-to-noise ratio, and
Phospholinked nucleotides, which produce a completely natural DNA strand through fast, accurate, and processive DNA synthesis.
DNA sequencing is performed on SMRT chips, each containing thousands of zero-mode waveguides (ZMWs). Utilizing the latest geometries available in semiconductor manufacturing, a ZMW is a hole, tens of nanometers in diameter, fabricated in a 100 nm metal film deposited on a silicon dioxide substrate. Each ZMW becomes a nanophotonic visualization chamber providing a detection volume of just 20 zeptoliters (10-21 liters). At this volume, the technology detects the activity of a single molecule among a background of thousands of labeled nucleotides.
The ZMW provides a window for watching DNA polymerase as it performs sequencing by synthesis. Within each chamber, a single DNA polymerase molecule is attached to the bottom surface such that it permanently resides within the detection volume. Phospholinked nucleotides, each type labeled with a different colored fluorophore, are then introduced into the reaction solution at high concentrations which promote enzyme speed, accuracy, and processivity. Due to the small size of the ZMW, even at these high biologically relevant concentrations, nucleotides occupy the detection volume only a small fraction of the time. In addition, visits to the detection volume are fast, lasting only a few microseconds due to the very small distance that diffusion has to carry the nucleotides. The result is a very low background.
DNA Sequencing Technologies
By: Jill U. Adams, Ph.D. (Freelance science writer in Albany, NY) © 2008 Nature Education ; Citation: Adams, J. (2008) DNA sequencing technologies. Nature Education 1.
The Human Genome project set out to sequence all of the 3 billion nucleotides in the human genome. Exactly how was this daunting task done with such incredible speed and accuracy?
The Human Genome Project set out to sequence the DNA of every human chromosome, thereby promising to advance knowledge of human biology and improve medicine. This project was huge in scale, as it sought to determine the order of all 3-3.2 billion nucleotides in the human genome. To reach this lofty goal, scientists developed a number of sequencing techniques that emphasized speed without too much loss of accuracy, heavily relying on automation and high-throughput technologies.
Early DNA Sequencing Technologies;
Early efforts at sequencing genes were painstaking, time-consuming, and labor-intensive, such as when Gilbert and Maxam reported the sequence of 24 base pairs using a method known as wandering-spot analysis (Gilbert & Maxam, 1973). Thankfully, this situation began to change during the mid-1970s, when researcher Frederick Sanger developed several faster, more efficient techniques to sequence DNA (Sanger et al., 1977). He sequenced the entire genome of phiX174 virus in five years (He took 10 yrs or so to sequence insulin amino acids). Indeed, Sanger's work in this area was so groundbreaking that it led to his receipt of the Nobel Prize in Chemistry in 1980.
The Rise of Industrial Sequencing Automation;
;)
In 1986, a company named Applied Biosystems began to manufacture automated DNA sequencing machines based on the Sanger method (Figure 1). These machines used fluorescent dyes to tag each nucleotide, allowing the reactions to be run in one column and read by color (rather than by what lane they appeared in). By running 24 samples at a time, the $100,000 machines yielded 12,000 "letters" of DNA per day. Although the machines were more expensive than traditional glass plates and gels, once the investment was made, these machines provided sequence data cheaper and faster than the traditional method. In fact, these are the machines that Craig Venter used to rally the Human Genome Project into high gear.
Today's machines can typically handle 96 samples at a time. In addition, whereas traditional, gel-based Sanger sequencing and early ABI machines could only generate 250-500 base pairs of DNA sequence per reaction; now 750-1000 base pairs of sequence can be read from a single reaction, making sequencing a much less expensive option than it used to be. In addition, sequencing technologies that don't use gels have been developed to further increase the efficiency of sequencing. These include flow cytometry, scanning microscopes, mass spectrometry, and hybridization strategies.
· High-throughput sequencing (WIKIPEDIA)
5.1 Lynx Therapeutics' Massively Parallel Signature Sequencing (MPSS) , 5.2 Polony sequencing, 5.3 454 pyrosequencing; 5.4 Illumina (Solexa) sequencing ; 5.5 SOLiD sequencing; 5.6 Ion semiconductor sequencing ; 5.7 DNA nanoball sequencing; 5.8 Helioscope(TM) single molecule sequencing ; 5.9 Single Molecule SMRT(TM) sequencing; 5.10 Single Molecule real time (RNAP) sequencing; 5.11 Nanopore DNA sequencing; 5.12 VisiGen Biotechnologies approach; 6 Future methods ; 7 Major landmarks in DNA sequencing. references See also references
High-throughput sequencing
The high demand for low-cost sequencing has driven the development of high-throughput sequencing technologies that parallelize the sequencing process, producing thousands or millions of sequences at once. High-throughput sequencing technologies are intended to lower the cost of DNA sequencing beyond what is possible with standard dye-terminator methods.
Lynx Therapeutics' Massively Parallel Signature Sequencing (MPSS):
Main article: Massively parallel signature sequencing.
The first of the "next-generation" sequencing technologies, MPSS was developed in the 1990s at Lynx Therapeutics, a company founded in 1992 by Sydney Brenner and Sam Eletr. MPSS was a bead-based method that used a complex approach of adapter ligation followed by adapter decoding, reading the sequence in increments of four nucleotides; this method made it susceptible to sequence-specific bias or loss of specific sequences. Because the technology was so complex, MPSS was only performed 'in-house' by Lynx Therapeutics and no machines were sold; when the merger with Solexa later led to the development of sequencing-by-synthesis, a more simple approach with numerous advantages, MPSS became obsolete. However, the essential properties of the MPSS output were typical of later "next-gen" data types, including hundreds of thousands of short DNA sequences. In the case of MPSS, these were typically used for sequencing cDNA for measurements of gene expression levels. Lynx Therapeutics merged with Solexa in 2004 and this company was later purchased by Illumina.
Polony sequencing
Polony sequencing, developed in the laboratory of George Church at Harvard, was among the first next-generation sequencing systems used to sequence a full genome in 2005. It combined an in vitro paired-tag library with emulsion PCR, an automated microscope, and ligation-based sequencing chemistry to sequence an E. coli genome at an accuracy of > 99.9999% and a cost approximately 1/10 that of Sanger sequencing. The technology was licensed to Agencourt Biosciences, subsequently spun out into Agencourt Personal Genomics, and ultimately incorporated into the Applied Biosystems SOLiD platform.
454 pyrosequencing:
Scitable by Nature Education
Main article: 454 Life Sciences Technology.
A parallelized version of pyrosequencing was developed by 454 Life Sciences, which has since been acquired by Roche Diagnostics. The method amplifies DNA inside water droplets in an oil solution (emulsion PCR), with each droplet containing a single DNA template attached to a single primer-coated bead that then forms a clonal colony. The sequencing machine contains many picolitre-volume wells each containing a single bead and sequencing enzymes. Pyrosequencing uses luciferase to generate light for detection of the individual nucleotides added to the nascent DNA, and the combined data are used to generate sequence read-outs. This technology provides intermediate read length and price per base compared to Sanger sequencing on one end and Solexa and SOLiD on the other.
Today, researchers are increasingly using a newer, single-nucleotide addition (SNA) method of DNA sequencing called pyrosequencing (Hyman, 1988) or 454 sequencing (after the name of the Roche-owned company that developed it). In pyrosequencing, the number of individual nucleotides is limited to the point at which DNA synthesis pauses. Then, unlike with the Sanger method, chain elongation can be resumed with the addition of nucleotides. A tiny amount of visible light is generated by enzymatic action as each nucleotide is added to a growing chain; this light is recorded as a series of peaks called a pyrogram, which corresponds to the order of lettered nucleotides that are added and ultimately reveals the underlying DNA sequence (Metzker, 2005). Thus, by correlating when a sample flashes with the nucleotide that is present at that time, researchers can sequence a stretch of DNA (Figure 3).
In its commercial version, 454 sequencing can read up to 20 million bases per run by applying the pyrosequencing technique on picolitre plates that facilitate sequencing of large amounts of DNA at low cost compared to earlier methods. Because the method does not rely on cloning template DNA, the read is more consistent (that is, it doesn't skip unclonable segments such as heterochromatin). However, one major drawback to the pyrosequencing approach is incomplete extension through homo-polymers, or simple repeats of the same nucleotide (e.g., AAAAAAA). Each read is only about 100 base pairs long at this time, making it difficult for scientists to differentiate between repeated regions longer than this length. However, pyrosequencing is improving quickly, and new machines can generate 400-base pair sequence reads.

a) Schematic of cyclic-array sequencing-by-synthesis methods (for example, fluorescent in situ sequencing, pyrosequencing, or single-molecule methods). Left: repeated cycles of polymerase extension with a single nucleotide at each step. The means of detecting incorporation events at individual array features varies from method to method. The sequencing templates shown here have been produced by using the POLONY method. Right: an example of raw data from Pyrosequencing, a cyclic-array method. The identity of nucleotides used at each extension step is listed along the x-axis. The y-axis depicts the measured signal at each cycle for one sequence; both single and multiple (such as homopolymeric) incorporations can be distinguished from non-incorporation events. The decoded sequence is listed along the top.
b) Sequencing by hybridization: To resequence a given base, four features are present on the microarray, each identical except for a different nucleotide at the query position (the central base of 25-bp oligonucleotides). Genotyping data at each base are obtained through the differential hybridization of genomic DNA to each set of four features. Copyright 2004 Nature publishing group, Shendure I., et. al., Advanced sequencing Technologies: methods and goals, Nature Reviews genetics 5, 335-344.
Illumina (Solexa) sequencing
Solexa, now part of Illumina, developed a sequencing technology based on reversible dye-terminators. DNA molecules are first attached to primers on a slide and amplified so that local clonal colonies are formed (bridge amplification). Four types of ddNTPs are added, and non-incorporated nucleotides are washed away. Unlike pyrosequencing, the DNA can only be extended one nucleotide at a time. A camera takes images of the fluorescently labeled nucleotides, then the dye along with the terminal 3' blocker is chemically removed from the DNA, allowing the next cycle.
SOLiD sequencing
Main article: ABI Solid Sequencing
Applied Biosystems' SOLiD technology employs sequencing by ligation. Here, a pool of all possible oligonucleotides of a fixed length is labeled according to the sequenced position. Oligonucleotides are annealed and ligated; the preferential ligation by DNA ligase for matching sequences results in a signal informative of the nucleotide at that position. Before sequencing, the DNA is amplified by emulsion PCR. The resulting bead, each containing only one copy of the same DNA molecule is deposited on a glass slide. The result is sequences of quantities and lengths comparable to Illumina sequencing.
Ion semiconductor sequencing:
Main article: Ion semiconductor sequencing
Ion Torrent Systems Inc. developed a system based on using standard sequencing chemistry, but with a novel, semiconductor based detection system. This method of sequencing is based on the detection of hydrogen ions that are released during the polymerization of DNA, as opposed to the optical methods used in other sequencing systems. A micro well containing a template DNA strand to be sequenced is flooded with a single type of nucleotide. If the introduced nucleotide is complementary to the leading template nucleotide it is incorporated into the growing complementary strand. This causes the release of a hydrogen ion that triggers a hypersensitive ion sensor, which indicates that a reaction has occurred. If homopolymer repeats are present in the template sequence multiple nucleotides will be incorporated in a single cycle. This leads to a corresponding number of released hydrogen and a proportionally higher electronic signal.
DNA nanoball sequencing
Main article: DNA nanoball sequencing
DNA nanoball sequencing is a type of high throughput sequencing technology used to determine the entire genomic sequence of an organism. The company Complete Genomics uses this technology to sequence samples that researchers submit from several projects. The method uses rolling circle replication to amplify small fragments of genomic DNA into DNA nanoballs. Unchained sequencing by ligation is then used to determine the nucleotide sequence. This method of DNA sequencing allows large numbers of DNA nanoballs to be sequenced per run and at low reagent costs compared to other next generation sequencing platforms. However, only short sequences of DNA are determined from each DNA nanoball which makes mapping the short reads to a reference genome difficult. This technology has been used for multiple genome sequencing projects and is scheduled to be used for more.
Helioscope(TM) single molecule sequencing
Main article: Helioscope (TM) single molecule sequencing
Based on "true single molecule sequencing" technology, Helioscope sequencing uses DNA fragments with added polyA tail adapters, which are attached to the flow cell surface. The next steps involve extension-based sequencing with cyclic washes of the flow cell with fluorescently labeled nucleotides (one nucleotide type at a time, as with the Sanger method). The reads are performed by the Helioscope sequencer. The reads are short, up to 55 bases per run, but recent improvement of the methodology allows more accurate reads of homopolymers (stretches of one type of nucleotides) and RNA sequencing.
Single Molecule SMRT(TM) sequencing
Main article: Single molecule SMRT (TM) sequencing
SMRT sequencing is based on the sequencing by synthesis approach. The DNA is synthesized in so called zero-mode wave-guides (ZMWs) - small well-like containers with the capturing tools located at the bottom of the well. The sequencing is performed with use of unmodified polymerase (attached to the ZMW bottom) and fluorescently labeled nucleotides flowing freely in the solution. The wells are constructed in a way that only the fluorescence occurring by the bottom of the well is detected. The fluorescent label is detached from the nucleotide at its incorporation into the DNA strand, leaving an unmodified DNA strand. According to Pacific Biosciences, the SMTR technology developer, this methodology allows detection of nucleotide modifications (such as cytosine methylation). This happens through the observation of polymerase kinetics. This approach allows reads of 1000 nucleotides.
Single Molecule real time (RNAP) sequencing
Main article: Single Molecule real time (RNAP) sequencing
This method is based on RNA polymerase (RNAP), which is attached to a polystyrene bead, with distal end of sequenced DNA is attached to another bead, with both beads being placed in optical traps. RNAP motion during transcription brings the beads in closer and their relative distance changes, which can then be recorded at a single nucleotide resolution. The sequence is deduced based on the four readouts with lowered concentrations of each of the four nucleotide types (similarly to Sanger’s method).
Nanopore DNA sequencing
Main article: Nanopore sequencing
This method is based on the readout of electrical signal occurring at nucleotides passing by alpha-hemolysin pores covalently bound with cyclodextrin. The DNA passing through the nanopore changes its ion current. This change is dependent on the shape, size and length of the DNA sequence. Each type of the nucleotide blocks the ion flow through the pore for a different period of time. The method has a potential of development as it does not require modified nucleotides, however single nucleotide resolution is not yet available.
VisiGen Biotechnologies approach
Main article: VisiGen Biotechnologies approach.
VisiGen Biotechnologies introduced a specially engineered DNA polymerase for use in their sequencing. This polymerase acts as a sensor - having incorporated a donor fluorescent dye by its active centre. This donor dye acts by FRET (fluorescent resonant energy transfer), inducing fluorescence of differently labeled nucleotides. This approach allows reads performed at the speed at which polymerase incorporates nucleotides into the sequence (several hundred per second). The nucleotide fluorochrome is released after the incorporation into the DNA strand. The expected read lengths in this approach should reach 1000 nucleotides, however this will have to be confirmed.
Future methods:
Sequencing by hybridization is a non-enzymatic method that uses a DNA microarray. A single pool of DNA whose sequence is to be determined is fluorescently labeled and hybridized to an array containing known sequences. Strong hybridization signals from a given spot on the array identifies its sequence in the DNA being sequenced. Mass spectrometry may be used to determine mass differences between DNA fragments produced in chain-termination reactions.
DNA sequencing methods currently under development include labeling the DNA polymerase, reading the sequence as a DNA strand transits through nanopores, and microscopy-based techniques, such as AFM or transmission electron microscopy that are used to identify the positions of individual nucleotides within long DNA fragments (>5,000 bp) by nucleotide labeling with heavier elements (e.g., halogens) for visual detection and recording. Third generation technologies aim to increase throughput and decrease the time to result and cost by eliminating the need for excessive reagents and harnessing the processivity of DNA polymerase.
In microfluidic Sanger sequencing the entire thermo cycling amplification of DNA fragments as well as their separation by electrophoresis is done on a single glass wafer (approximately 10 cm in diameter) thus reducing the reagent usage as well as cost. In some instances researchers have shown that they can increase the throughput of conventional sequencing through the use of microchips. Research will still need to be done in order to make this use of technology effective.
In October 2006, the X Prize Foundation established an initiative to promote the development of full genome sequencing technologies, called the Archon X Prize, intending to award $10 million to "the first Team that can build a device and use it to sequence 100 human genomes within 10 days or less, with an accuracy of no more than one error in every 100,000 bases sequenced, with sequences accurately covering at least 98% of the genome, and at a recurring cost of no more than $10,000 (US) per genome.
Each year NHGRI promotes grants for new research and developments in genomics. 2010 grants and 2011 candidates include continuing work in microfluidic, Polony and base-heavy sequencing methodologies.
New DNA Sequencing Technology Uses Firefly Enzymes To Read Genetic Code; Science Daily (July 12, 2007) — Unique technology that uses the enzymes of fireflies to read the genetic code of DNA has been installed at the University of Liverpool.
Liverpool is one of only two universities in the UK with the machine, which can read up to 100 million DNA letters in a few hours compared to technology currently in use that can only process 50,000. The machine - called GS-Flex - is unique in that it uses an enzyme found in fireflies as a flash light to help read the DNA strand.
Faster DNA Sequencing, Tuesday, April 8, 2008
By Emily Singer
New techniques could finally make a $1,000 genome possible.
Helicos launched its commercial technology--the first to use single molecules--last month. Other companies are already nipping at Helicos' heels, with promises to deliver faster and cheaper technologies within the next few years. But it's not yet clear what the best approach will be or how long it will take to reach the $1,000 goal, a price thought to reflect how much the average American could afford to pay for a once-in-a-lifetime medical service. Scientists hope that genome sequencing will ultimately become an integral part of an individual's medical record, helping to determine a person's risk of acquiring specific diseases, as well as the best-suited treatments. In the last few years, gene sequencing technologies have dropped exponentially in cost from $3 billion for the Human Genome Project to less than $100,000, according to recent announcements from genomics companies Illumina and Applied Biosciences. The next-generation sequencing technologies currently in use, including those from Illumina, Applied Biosystems, and 454, require that the DNA molecule be amplified many times and then read simultaneously, making it easier to detect the fluorescent markers that indicate the position of each DNA letter. But single molecule sequencing gets rid of the amplification step, greatly simplifying the process.
The company's sequencing-by-synthesis approach is similar to that used by other machines--DNA molecules are chopped into smaller fragments and attached to a slide, which is flooded with fluorescently labeled bases or DNA letters. A camera captures the series of signals that results when an enzyme attaches the appropriate base to the attached piece of DNA. The Helicos device, however, can read the signal from a single molecule, rather than the thousands needed for other machines.
Key Advance In DNA Sequencing With Nanopores; High-quality detection takes the label-free, single-molecule technique closer to reality;
Stu Borman
TAKING DNA APART one nucleotide at a time, directing the nucleotides sequentially into a nanopore and detecting them with an electrical current meter may seem an unlikely DNA-sequencing concept, but it is closer than ever to being a reality.
Chemical biology professor Hagan Bayley of the University of Oxford and coworkers there and at Oxford Nanopore Technologies, a company Bayley founded, have just demonstrated that the four standard DNA nucleotides—adenine, thymine, guanine, and cytosine—can be distinguished from one another reliably by the amount of current they each block as they flow through a Nanopore (Nat. Nanotechnology., DOI: 10.1038/nnano.2009.12). And for good measure, they've shown that the method can also identify 5-methylcytosine, a biologically important modified nucleotide.
A key advantage of nanopore sequencing is that it detects nucleotides directly, without reagents or labeling, from a single molecule of single-stranded DNA. All current DNA-sequencing techniques require fluorescent or luminescent labeling, and most use polymerase chain reaction for DNA amplification. Nanopore technology is also potentially capable of sequencing much longer continuous strands of DNA than other techniques. This capability is becoming increasingly important for studies of long-range genomic complexity to associate DNA rearrangements with cancer and other diseases.
EVENTUAL GOAL Nanopore DNA sequencing, as envisioned, will use an exonuclease (green) to cleave nucleotides from DNA. Each nucleotide will be directed into the nanopore (blue), where it interacts with a cyclodextrin "adapter" (red, donut-shaped) and interrupts a current to an extent that pinpoints its identity (such as G = guanine, C = cytosine, and T = thymine).
National Human Genome Research Institute at the National Institutes of Health, says Bayley and coworkers "showed that one can operate the system under conditions where an exonuclease can function and still get the released nucleotides to pass through the pore and distinguish them. Daniel Branton of Harvard University and Stuart Lindsay of Arizona State University, an expert in single-molecule DNA sequencing, says Bayley and coworkers "certainly have a wonderful single-nucleotide detector technology.

Ion Semiconductor Sequencing 2010.. (WIKIPedia) This is a method of DNA sequencing based on the detection of hydrogen ions that are released during the polymerization of DNA. This is a method of "sequencing by synthesis", during which a complementary strand is built based on the sequence of a template stand.
A microwell containing a template DNA strand to be sequenced is flooded with a single species of deoxy ribonucleotide (dNTP). If the introduced dNTP is complementary to the leading template nucleotide, it is incorporated into the growing complementary strand. This causes the release of a hydrogen ion that triggers a hypersensitive ion sensor, which indicates that a reaction has occurred. If homopolymer repeats are present in the template sequence, multiple dNTP molecules will be incorporated in a single cycle. This leads to a corresponding number of released hydrogen and a proportionally higher electronic signal.
This technology differs from other sequencing technologies in that no modified nucleotides or optics are used. Ion semiconductor sequencing may also be referred to as ion torrent sequencing, pH-mediated sequencing, silicon sequencing, or semiconductor sequencing. It was developed by Ion Torrent Systems Inc. and was released in February 2010. Ion Torrent have marketed their machine as a rapid, compact and economical sequencer that can be utilized in a large number of laboratories as a bench top machine.[2
New DNA sequencing cheap as chips; Thursday, 21 July 2011; by Claire MacDonald. [Face Book].
Fast, cheap sequencing.
Jonathan Rothberg and colleagues at Life Technologies Ion Torrent in California have developed a solution to this problem in the form of an electronic sensor in an integrated circuit that is produced using low-cost semiconductor manufacturing techniques.
"The development of ion semiconductor sequencing will have as profound an effect on sequencing as the introduction of CMOS imagers had on the development of digital photography - it will make sequencing ubiquitous, fast and low cost," said Rothberg, who has described the new technology in an article published in Nature this week.
"Genome sequencing has huge potential in the diagnosis and management of disease. This semiconductor device is a very exciting development that should be able to deliver genome information to the clinic cheaply and effectively," added Dame Kay Davies from the University of Oxford, UK, who was not involved in the research.
A novel approach:
Current DNA sequencing methods use optical imaging, which requires costly equipment, while other attempts to use electronic sensors to synthesize and sequence DNA have been limited by the number of sensors that could fit on a single array, which adversely affects the speed at which they can read the code.
Rothberg's team has overcome these limitations by producing simple electronic chips with a large number of fast, uniform sensors arranged in a two-dimensional array.
Unlike the current light-based sequencing techniques, which rely on the release of photons to measure when a nucleotide base has been incorporated into a DNA strand, Ion Torrent's chips have no optical components and instead use a sensor to measure the release of hydrogen ions, which cause a reduction in pH to the surrounding medium.
This pH change is converted to a voltage when each nucleotide base is incorporated, so that the code of the DNA strand can be read from the series of voltage readings by a signal processor. Each nucleotide base pair is flowed sequentially over the chip to differentiate between the four nucleotides. The results are passed through several filters to ensure that the ‘read’ of the DNA is accurate and any erroneous results are filtered out.
Sequencing Chemistry
In nature, the incorporation of a deoxyribonucleotides (dNTP) into a growing DNA strand involves the formation of a covalent bond and the release of pyrophosphate and a positively charged hydrogen ion.[1] A dNTP will only be incorporated if it is complementary to the leading unpaired template nucleotide. Ion semiconductor sequencing exploits these facts by determining if a hydrogen ion is released upon providing a single species of dNTP to the reaction.
Micro wells on a semiconductor chip that each contain one single-stranded template DNA molecule to be sequenced and one DNA polymerase are sequentially flooded with unmodified A, C, G or T dNTP. If an introduced dNTP is complementary to the next unpaired nucleotide on the template strand it is incorporated into the growing complementary strand by the DNA polymerase. If the introduced dNTP is not complementary there is no incorporation and no biochemical reaction. The hydrogen ion that is released in the reaction changes the pH of the solution, which is detected by a hypersensitive ion sensor. The unattached dNTP molecules are washed out before the next cycle when a different dNTP species is introduced.
Signal Detection
Beneath the layer of microwells is an ion sensitive layer, below which is a hypersensitive ISFET ion sensor. All layers are contained within a CMOS semiconductor chip, similar to that used in the electronics industry.
Each released hydrogen ion triggers the ISFET ion sensor. The series of electrical pulses transmitted from the chip to a computer is translated into a DNA sequence, with no intermediate signal conversion required. Each chip contains an array of microwells with corresponding ISFET detectors.[3] Because nucleotide incorporation events are measured directly by electronics, the use of labeled nucleotides and optical measurements are avoided.
Sequencing Characteristics
The per base accuracy achieved in house by Ion Torrent on the Ion Torrent ion semiconductor sequencer as of February 2011 was 99.6% based on 50 base reads, with 100 Mb per run. The read-length as of February 2011 was 100 base pairs. The accuracy for homopolymer repeats of 5 repeats in length was 98%.. It should be noted that these figures have not yet been independently verified outside of the company.

The release of hydrogen ions indicate if zero, one or more nucleotides were incorporated.

Released hydrogen ions are detected by an ion sensor. Multiple incorporations lead to a corresponding number of released hydrogen and intensity of signal.
BioMed Central Blog:
Latest advances in DNA sequencing technology; Investigative Genetics: There has been a rapid evolution of Second Generation Sequencing (SGS) techniques. Second Generation Sequencing (SGS) techniques. These methods are able to provide a much higher throughput, and therefore a lower cost per sequenced base. Investigative Genetics publishes articles on the development and application of molecular genetics in a wide range of science disciplines with societal relevance.
The National Human Genome Research Institute (NHGRI) Genome Technology Program Overview (October 3rd 2011).
The Genome Technology program supports research to develop new methods, technologies and instruments that enable rapid, low-cost determination of DNA sequence, SNP genotyping (Genetic Variation Program) and functional genomics (broadly defined) experiments (Functional Analysis Program). Priorities include the refinement of current technologies to increase efficiency and decrease cost while maintaining or improving data quality, and the development of completely novel approaches to achieve orders-of-magnitude improvement. Integration of process steps is key to achieving these goals.
The program also supports and coordinates transfer of technology from developers to users, and promotes collaborative, multidisciplinary programs that closely integrate research projects at academic and industrial laboratories.
The National Human Genome Research Institute (NHGRI) participates in numerous trans-NIH efforts, including one on bioengineering research. The development of integrated concepts and tools for genomic research benefits from bioengineering research, methods and approaches. NHGRI supports bioengineering research through grants made in response to NHGRI program announcements and requests for application, and through NHGRI participation in trans-NIH bioengineering program announcements, such as the Bioengineering Research Grants (BRG) and Bioengineering Research Partnerships (BRP) programs and other Funding Opportunities that are listed below.
Epoch Life Science provides (21 November 2011) a next-generation sequencing service using Ion Torrent Personal Genome Machine (PGM). The PGM™ sequencer is a bench top system utilizing semiconductor technology that enables rapid and scalable sequencing experiments.
Ion Torrent technology uses a massively parallel array of proprietary semiconductor sensors to perform direct real time measurement of the hydrogen ions produced during DNA replication. A high-density array of wells on the Ion semiconductor chips provide millions of individual reactors while integrated fluidics allows reagents to flow over the sensor array. This unique combination of fluidics, micromachining, and semiconductor technology enable the direct translation of genetic information (DNA) to digital information (DNA sequence) rapidly and generating >10Mb (314 chip) and >100Mb (316 chip) data output respectively. A typical sequencing run takes only 2-3 hours. Project pipeline is as below.
Major landmarks in DNA sequencing
· 1953 Discovery of the structure of the DNA double helix.[48]
· 1972 Development of recombinant DNA technology, which permits isolation of defined fragments of DNA; prior to this, the only accessible samples for sequencing were from bacteriophage or virus DNA.
· 1977 The first complete DNA genome to be sequenced is that of bacteriophage φX174.[49]
· 1977 Allan Maxam and Walter Gilbert publish "DNA sequencing by chemical degradation".[5] Frederick Sanger, independently, publishes "DNA sequencing with chain-terminating inhibitors".[9]
· 1984 Medical Research Council scientists decipher the complete DNA sequence of the Epstein-Barr virus, 170 kb.
· 1986 Leroy E. Hood's laboratory at the California Institute of Technology and Smith announce the first semi-automated DNA sequencing machine.
· 1987 Applied Biosystems markets first automated sequencing machine, the model ABI 370.
· 1990 The U.S. National Institutes of Health (NIH) begins large-scale sequencing trials on Mycoplasma capricolum, Escherichia coli, Caenorhabditis elegans, and Saccharomyces cerevisiae (at US$0.75/base).
· 1991 Sequencing of human expressed sequence tags begins in Craig Venter's lab, an attempt to capture the coding fraction of the human genome.[50]
· 1995 Craig Venter, Hamilton Smith, and colleagues at The Institute for Genomic Research (TIGR) publish the first complete genome of a free-living organism, the bacterium Haemophilus influenzae. The circular chromosome contains 1,830,137 bases and its publication in the journal Science[51] marks the first use of whole-genome shotgun sequencing, eliminating the need for initial mapping efforts.
· 1996 Pål Nyrén and his student Mostafa Ronaghi at the Royal Institute of Technology in Stockholm publish their method of pyrosequencing[52]
·
1998 Phil Green and Brent Ewing of the
University of Washington publish "phred" for sequencer data
analysis.[53]
· 2000 Lynx Therapeutics publishes and markets "MPSS" - a parallelized, adapter/ligation-mediated, bead-based sequencing technology, launching "next-generation" sequencing.[54]
- 2000 Celera and collaborators sequence fruit fly Drosophila melanogaster (genome size of 180Mb) - validation of Venter's shotgun method. HGP and Celera debate issues related to data release.
- HGP consortium publishes sequence of chromosome 21.[42]
- HGP & Celera jointly announce working drafts of HG sequence, promise joint publication.
- Estimates for the number of genes in the human genome range from 35,000 to 120,000. International consortium completes first plant sequence, Arabidopsis thaliana (genome size of 125 Mb).
·
· 2001 A draft sequence of the human genome is published.[55][56]
- 2001 HGP consortium publishes Human Genome Sequence draft in Nature (15 Feb)[43].
- Celera publishes the Human Genome sequence[44].
·
· 2004 454 Life Sciences markets a parallelized version of pyrosequencing.[57][58] The first version of their machine reduced sequencing costs 6-fold compared to automated Sanger sequencing, and was the second of a new generation of sequencing technologies, after MPSS.[29]
· 2005 420,000 VariantSEQr human resequencing primer sequences published on new NCBI Probe database.
· 2007 For the first time, a set of closely related species (12 Drosophilidae) are sequenced, launching the era of phylogenomics.
o Craig Venter publishes his full diploid genome: the first human genome to be sequenced completely.
DNA Microarray Methods:
Comparative gene expression profiling employs the use of DNA microarrays, often referred to as DNA “chips.”. Two basic types of DNA microarrays are currently available: oligonucleotide arrays and cDNA arrays. Both approaches involve the immobilization of DNA sequences in a gridded array on the surface of a solid support, such as a glass microscope slide or silicon wafer. In the case of oligonucleotide arrays, 25-nucleotide long fragments of known DNA sequence are synthesized in situ on the surface of the chip using a series of light-directed coupling reactions similar to photolithography. Using this method, as many as 300,000 distinct sequences representing over 6,000 genes can be synthesized on a single 1.3 cm × 1.3 cm microarray. In the case of cDNA microarrays, cDNA fragments are deposited onto the surface of a glass slide using a robotic spotting device. For both microarray approaches, the next step involves the purification of RNA from the source of interest (e.g., from a tumor), enzymatic fluorescent labeling of the RNA, and hybridization of the fluorescently labeled material to the microarray. Hybridization events are then captured by scanning the surface of the microarray with a laser scanning device and measuring the fluorescence intensity at each position in the microarray. The fluorescence intensity of each spot on the array is proportional to the level of expression of the gene represented by that spot. This process is illustrated( BIORAD- quantification of Gene expression)
DNA oligos representing each of the genes are plated on to microchips or they are synthesized on the plate made up of glass plates or membrane. The oligos are spotted and numbered and each of the spots contains several thousands of oligos representing specific genes. Each spot may contain thousands of similar oligos of 30-50 nucleotides long. In order to produce such oligos one has to have knowledge of the genomic sequence or expressed CDNA sequence.
To find out the genes expressed in different tissue and quantify the amount of mRNA produced in different tissues, isolate mRNA prepare cDNA with labeled color dyes and hybridize and find out the which are genes expressed. The intensity of the color in a slot gives the quantity; the mRNAs expressed in different tissue can also be evaluated from the same microarray plate.
Spot the oligos of 25-30ntd long and fix to the silicon or glass plate. Note the horizontal and vertical rows and each spot represents. A single plate can have 1000 to 100 spots.
Wash the spots with suitable buffer. Prepare cDNA from the expressed tissue one and two with labeled compounds. Drop the labeled cDNA from tissue one; one should note the amount of cDNA used for every spot. Hybridize and remove the unhybridized cDNA and expose the plate to UV light. Wherever spots hybridized show Blue fluorescence for the label is blue emitting dye. Depending upon the intensity of light emitted one can calculate approximate amount of specific RNA expressed. Intense fluorescence in a spot suggests the quantity of the mRNA is high. Some spots show no fluorescence, it means the said gene is not expressed. The red labeled cDNA from another tissue when some spots show fluorescent orange color; it indicates the same gene is expressed in both types of tissue. The said spots indicate the mRNAs expressed are same. In some one finds only red; it means it is specific to second tissue. The intensity of the fluorescence can be used for quantitation.

DNA microarray
RNA extracted from a tissue is end-labeled with a fluorescent marker, then allowed to hybridize to a chip derivatized with cDNAs or oligonucleotides as described in the text. The precise location of RNA hybridization to the chip can be determined using a laser scanner. Since the position of each unique cDNA or oligonucleotide is known, the presence of a cognate RNA for any given unique sequence can be determined. http://www.transcriptome.ens.fr/